

Stratification of Wilms tumor by genetic and epigenetic analysis
- PMID: 22470196
- PMCID: PMC3359888
- DOI: 10.18632/oncotarget.468
Stratification of Wilms tumor by genetic and epigenetic analysis
Abstract
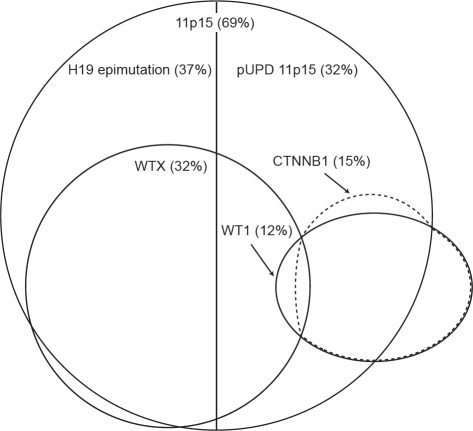
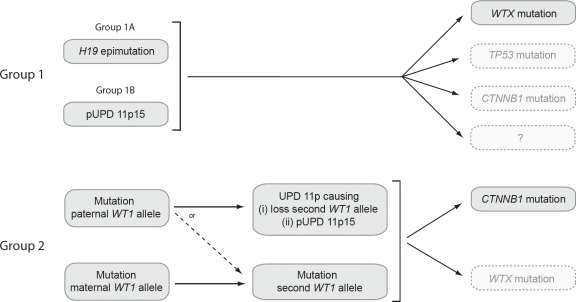
Somatic defects at five loci, WT1, CTNNB1, WTX, TP53 and the imprinted 11p15 region, are implicated in Wilms tumor, the commonest childhood kidney cancer. In this study we analysed all five loci in 120 Wilms tumors. We identified epigenetic 11p15 abnormalities in 69% of tumors, 37% were H19 epimutations and 32% were paternal uniparental disomy (pUPD). We identified mutations of WTX in 32%, CTNNB1 in 15%, WT1 in 12% and TP53 in 5% of tumors. We identified several significant associations: between 11p15 and WTX (P=0.007), between WT1 and CTNNB1 (P less than 0.001), between WT1 and pUPD 11p15 (P=0.01), and a strong negative association between WT1 and H19 epimutation (P less than 0.001). We next used these data to stratify Wilms tumor into three molecular Groups, based on the status at 11p15 and WT1. Group 1 tumors (63%) were defined as 11p15-mutant and WT1-normal; a third also had WTX mutations. Group 2 tumors (13%) were WT1-mutant. They either had 11p15 pUPD or were 11p15-normal. Almost all had CTNNB1 mutations but none had H19 epimutation. Group 3 tumors (25%) were defined as 11p15-normal and WT1-normal and were typically normal at all five loci (P less than 0.001). We also identified a novel clinical association between H19 epimutation and bilateral disease (P less than 0.001). These data provide new insights into the pattern, order, interactions and clinical associations of molecular events in Wilms tumor.
Figures
Similar articles
-
Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors.Genes Chromosomes Cancer. 2008 Jun;47(6):461-70. doi: 10.1002/gcc.20553. Genes Chromosomes Cancer. 2008. PMID: 18311776 Free PMC article.
-
WT1, WTX and CTNNB1 mutation analysis in 43 patients with sporadic Wilms' tumor.Oncol Rep. 2013 Jan;29(1):315-20. doi: 10.3892/or.2012.2096. Epub 2012 Oct 19. Oncol Rep. 2013. PMID: 23117548
-
Frequency of WT1 and 11p15 constitutional aberrations and phenotypic correlation in childhood Wilms tumour patients.Eur J Cancer. 2012 Nov;48(17):3249-56. doi: 10.1016/j.ejca.2012.06.008. Epub 2012 Jul 14. Eur J Cancer. 2012. PMID: 22796116
-
Pathology, genetics and cytogenetics of Wilms' tumour.Pathology. 2011 Jun;43(4):302-12. doi: 10.1097/PAT.0b013e3283463575. Pathology. 2011. PMID: 21516053 Review.
-
Wilms' tumours: about tumour suppressor genes, an oncogene and a chameleon gene.Nat Rev Cancer. 2011 Feb;11(2):111-21. doi: 10.1038/nrc3002. Epub 2011 Jan 20. Nat Rev Cancer. 2011. PMID: 21248786 Free PMC article. Review.
Cited by
-
Genes Controlled by DNA Methylation Are Involved in Wilms Tumor Progression.Cells. 2019 Aug 17;8(8):921. doi: 10.3390/cells8080921. Cells. 2019. PMID: 31426508 Free PMC article.
-
A high incidence of WT1 abnormality in bilateral Wilms tumours in Japan, and the penetrance rates in children with WT1 germline mutation.Br J Cancer. 2015 Mar 17;112(6):1121-33. doi: 10.1038/bjc.2015.13. Br J Cancer. 2015. PMID: 25688735 Free PMC article.
-
Nuclear β-Catenin Expression is Frequent in Sinonasal Hemangiopericytoma and Its Mimics.Head Neck Pathol. 2017 Jun;11(2):119-123. doi: 10.1007/s12105-016-0737-2. Epub 2016 Jun 20. Head Neck Pathol. 2017. PMID: 27325236 Free PMC article.
-
HnRNPL promotes Wilms tumor progression by regulating the p53 and Bcl2 pathways.Onco Targets Ther. 2019 May 29;12:4269-4279. doi: 10.2147/OTT.S203046. eCollection 2019. Onco Targets Ther. 2019. PMID: 31213844 Free PMC article.
-
Genetic and epigenetic features of bilateral Wilms tumor predisposition in patients from the Children's Oncology Group AREN18B5-Q.Nat Commun. 2023 Dec 18;14(1):8006. doi: 10.1038/s41467-023-43730-0. Nat Commun. 2023. PMID: 38110397 Free PMC article.
References
-
- Breslow NE, Olson J, Moksness J, Beckwith JB, Grundy P. Familial Wilms' tumor: a descriptive study. Med Pediatr Oncol. 1996;27:398–403. - PubMed
-
- Bardeesy N, Falkoff D, Petruzzi MJ, Nowak N, Zabel B, Adam M, Aguiar MC, Grundy P, Shows T, Pelletier J. Anaplastic Wilms' tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet. 1994;7:91–97. - PubMed
-
- Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell. 1990;60:509–520. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
