

E-cadherin destabilization accounts for the pathogenicity of missense mutations in hereditary diffuse gastric cancer
- PMID: 22470475
- PMCID: PMC3309996
- DOI: 10.1371/journal.pone.0033783
E-cadherin destabilization accounts for the pathogenicity of missense mutations in hereditary diffuse gastric cancer
Abstract
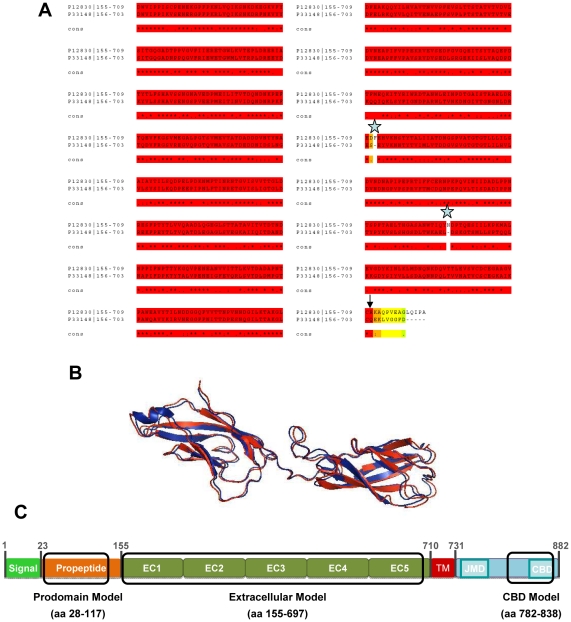
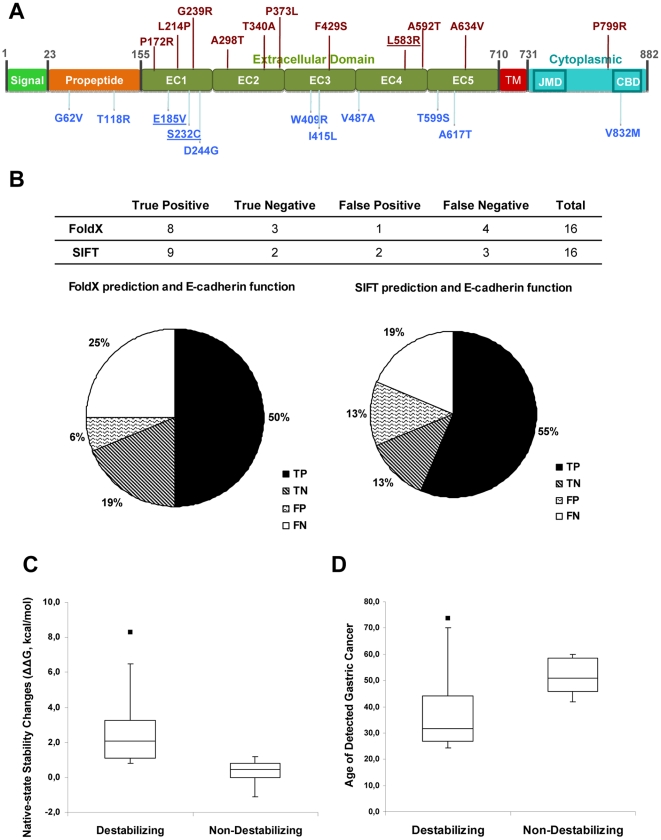
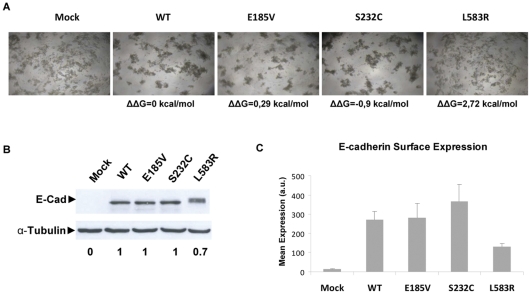
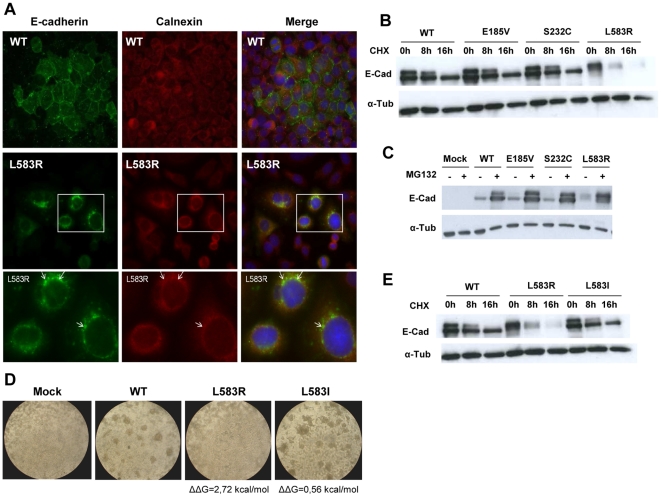
E-cadherin is critical for the maintenance of tissue architecture due to its role in cell-cell adhesion. E-cadherin mutations are the genetic cause of Hereditary Diffuse Gastric Cancer (HDGC) and missense mutations represent a clinical burden, due to the uncertainty of their pathogenic role. In vitro and in vivo, most mutations lead to loss-of-function, although the causal factor is unknown for the majority. We hypothesized that destabilization could account for the pathogenicity of E-cadherin missense mutations in HDGC, and tested our hypothesis using in silico and in vitro tools. FoldX algorithm was used to calculate the impact of each mutation in E-cadherin native-state stability, and the analysis was complemented with evolutionary conservation, by SIFT. Interestingly, HDGC patients harbouring germline E-cadherin destabilizing mutants present a younger age at diagnosis or death, suggesting that the loss of native-state stability of E-cadherin accounts for the disease phenotype. To elucidate the biological relevance of E-cadherin destabilization in HDGC, we investigated a group of newly identified HDGC-associated mutations (E185V, S232C and L583R), of which L583R is predicted to be destabilizing. We show that this mutation is not functional in vitro, exhibits shorter half-life and is unable to mature, due to premature proteasome-dependent degradation, a phenotype reverted by stabilization with the artificial mutation L583I (structurally tolerated). Herein we report E-cadherin structural models suitable to predict the impact of the majority of cancer-associated missense mutations and we show that E-cadherin destabilization leads to loss-of-function in vitro and increased pathogenicity in vivo.
Conflict of interest statement
Figures
Similar articles
-
The importance of E-cadherin binding partners to evaluate the pathogenicity of E-cadherin missense mutations associated to HDGC.Eur J Hum Genet. 2013 Mar;21(3):301-9. doi: 10.1038/ejhg.2012.159. Epub 2012 Aug 1. Eur J Hum Genet. 2013. PMID: 22850631 Free PMC article.
-
Identification of CDH1 germline missense mutations associated with functional inactivation of the E-cadherin protein in young gastric cancer probands.Hum Mol Genet. 2003 Mar 1;12(5):575-82. doi: 10.1093/hmg/ddg048. Hum Mol Genet. 2003. PMID: 12588804
-
Predicting the Functional Impact of CDH1 Missense Mutations in Hereditary Diffuse Gastric Cancer.Int J Mol Sci. 2017 Dec 12;18(12):2687. doi: 10.3390/ijms18122687. Int J Mol Sci. 2017. PMID: 29231860 Free PMC article. Review.
-
Characterization of the P373L E-cadherin germline missense mutation and implication for clinical management.Eur J Surg Oncol. 2007 Nov;33(9):1061-7. doi: 10.1016/j.ejso.2007.03.001. Epub 2007 Apr 16. Eur J Surg Oncol. 2007. PMID: 17434710
-
[From gene to disease; E-cadherin and hereditary diffuse gastric cancer].Ned Tijdschr Geneeskd. 2003 Dec 13;147(50):2474-7. Ned Tijdschr Geneeskd. 2003. PMID: 14708213 Review. Dutch.
Cited by
-
Geographical Distribution of E-cadherin Germline Mutations in the Context of Diffuse Gastric Cancer: A Systematic Review.Cancers (Basel). 2021 Mar 12;13(6):1269. doi: 10.3390/cancers13061269. Cancers (Basel). 2021. PMID: 33809393 Free PMC article. Review.
-
CDH1 Genotype Exploration in Women With Hereditary Lobular Breast Cancer Phenotype.JAMA Netw Open. 2024 Apr 1;7(4):e247862. doi: 10.1001/jamanetworkopen.2024.7862. JAMA Netw Open. 2024. PMID: 38652475 Free PMC article.
-
Comparative study of endoscopic surveillance in hereditary diffuse gastric cancer according to CDH1 mutation status.Gastrointest Endosc. 2018 Feb;87(2):408-418. doi: 10.1016/j.gie.2017.06.028. Epub 2017 Jul 6. Gastrointest Endosc. 2018. PMID: 28688938 Free PMC article.
-
Molecular chaperones: Guardians of tumor suppressor stability and function.Oncotarget. 2024 Oct 1;15:679-696. doi: 10.18632/oncotarget.28653. Oncotarget. 2024. PMID: 39352796 Free PMC article. Review.
-
Quantification of mutant E-cadherin using bioimaging analysis of in situ fluorescence microscopy. A new approach to CDH1 missense variants.Eur J Hum Genet. 2015 Aug;23(8):1072-9. doi: 10.1038/ejhg.2014.240. Epub 2014 Nov 12. Eur J Hum Genet. 2015. PMID: 25388006 Free PMC article.
References
-
- Hulpiau P, van Roy F. Molecular evolution of the cadherin superfamily. Int J Biochem Cell Biol. 2009;41:349–369. - PubMed
-
- Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta. 1994;1198:11–26. - PubMed
-
- Mareel M, Leroy A. Clinical, cellular, and molecular aspects of cancer invasion. Physiol Rev. 2003;83:337–376. - PubMed
-
- Berx G, Becker KF, Hofler H, van Roy F. Mutations of the human E-cadherin (CDH1) gene. Hum Mutat. 1998;12:226–237. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
