

Tumour necrosis factor-mediated macrophage activation in the target organ is critical for clinical manifestation of uveitis
- PMID: 22471277
- PMCID: PMC3390517
- DOI: 10.1111/j.1365-2249.2012.04567.x
Tumour necrosis factor-mediated macrophage activation in the target organ is critical for clinical manifestation of uveitis
Abstract
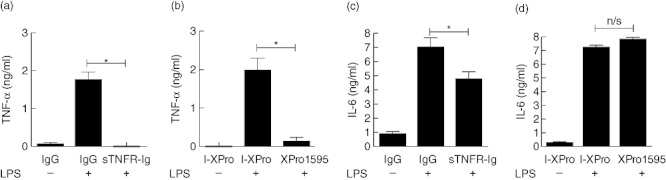
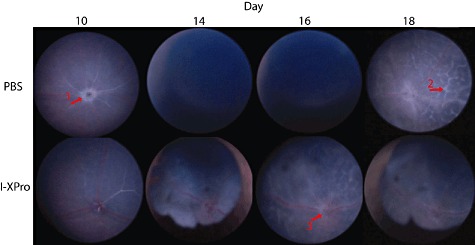
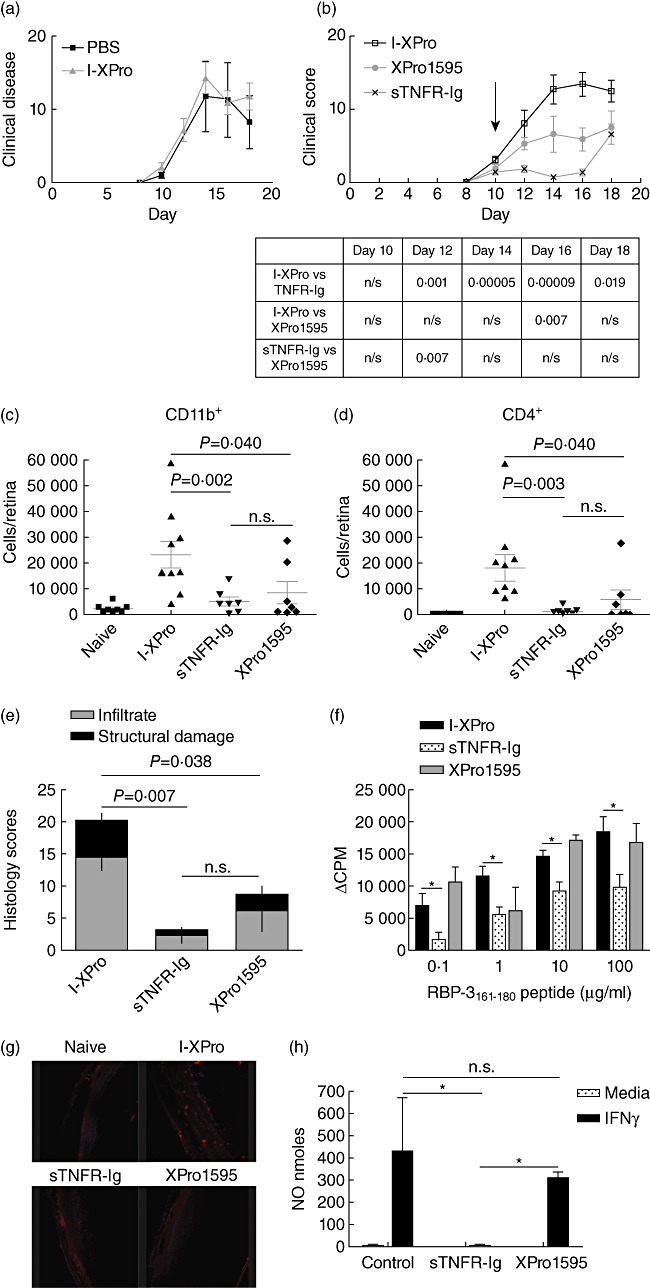
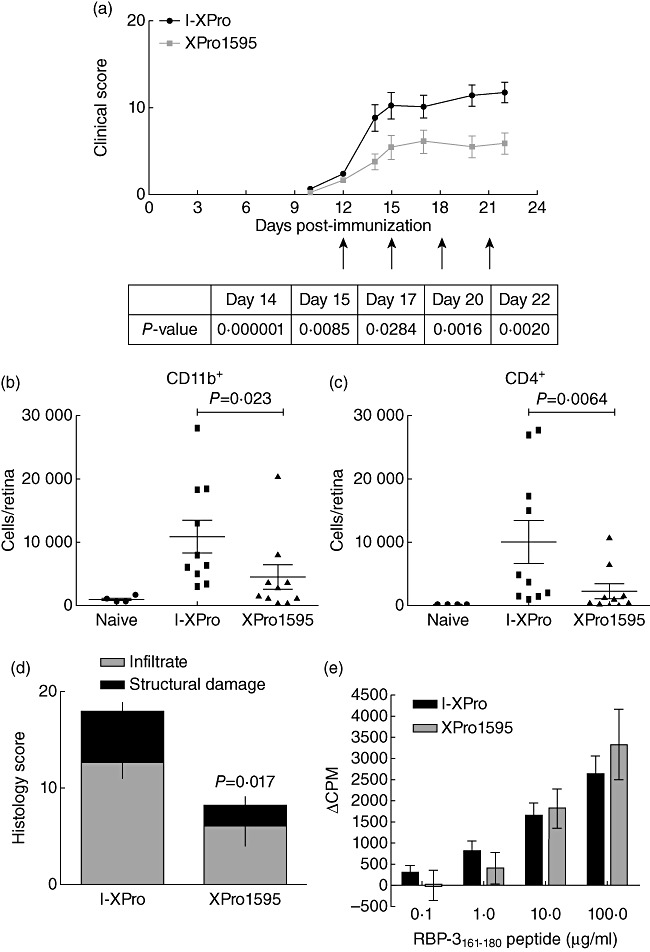
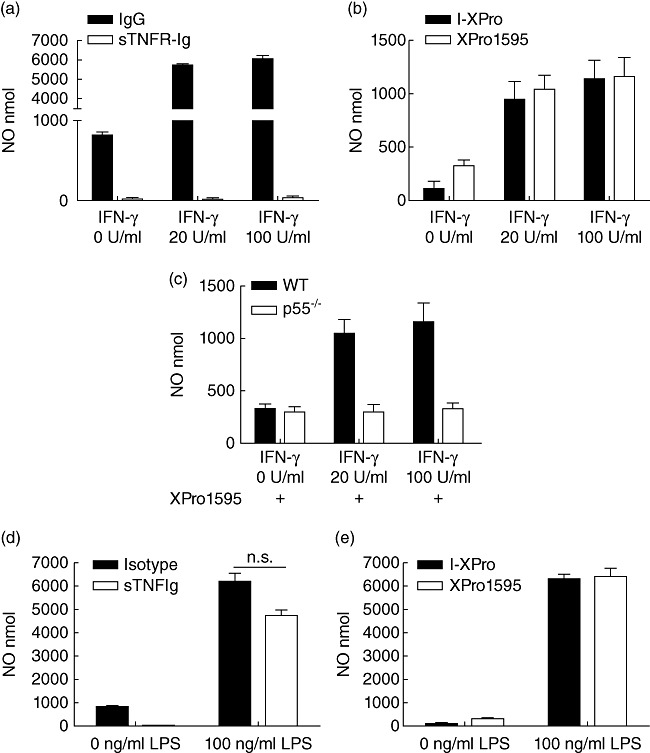
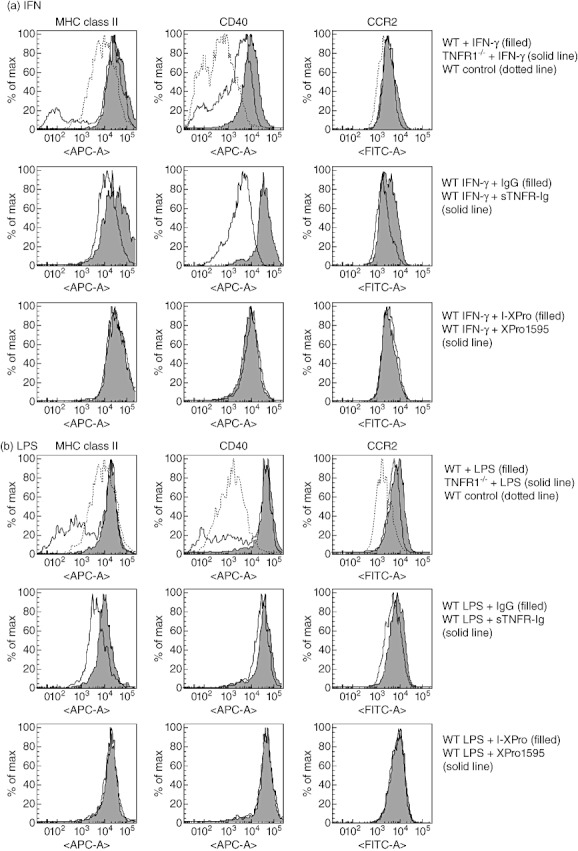
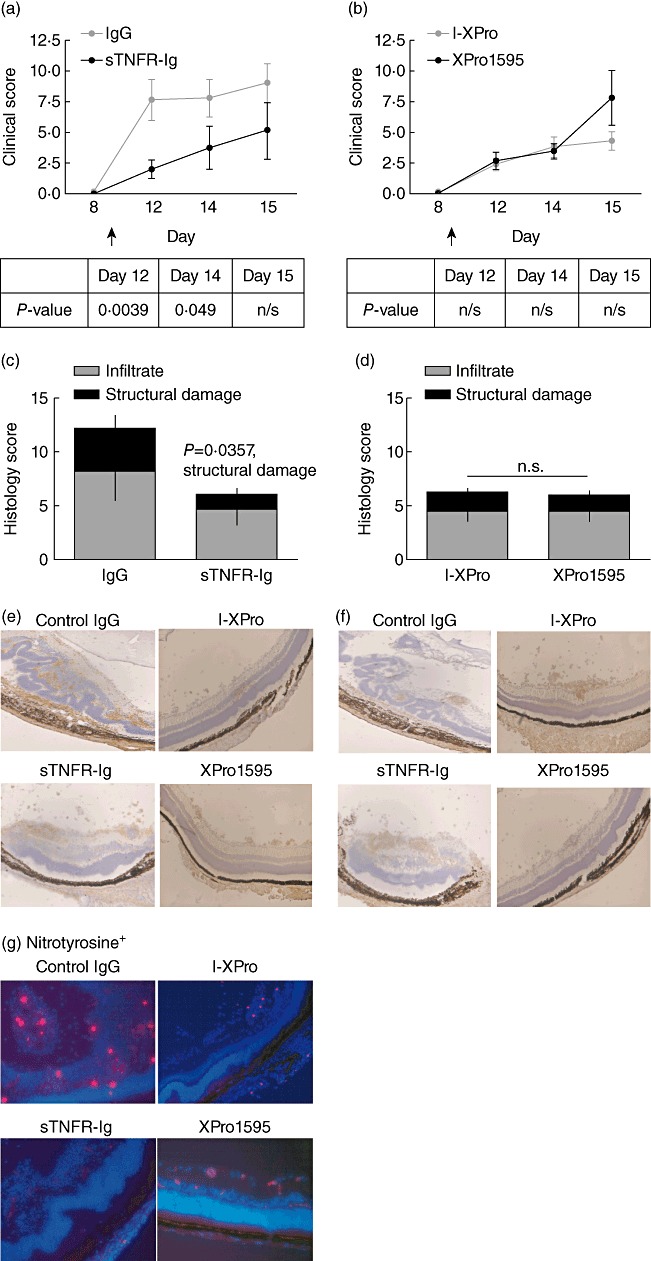
Clinically available anti-tumour necrosis factor (TNF) biologics, which inhibit both soluble (sTNF) and transmembrane forms (tmTNF) of TNF, eliminating all TNF signalling, have successfully treated autoimmune diseases including uveitis. These have potentially serious side effects such as reactivation of latent Mycobacterium tuberculosis and, therefore, more specific inhibition of TNF signalling pathways may maintain clinical efficacy while reducing adverse effects. To determine the effects of specific pharmacological inhibition of sTNF on macrophage activation and migration, we used a mouse model of uveitis (experimental autoimmune uveoretinitis; EAU). We show that selective inhibition of sTNF is sufficient to suppress EAU by limiting inflammatory CD11b(+) macrophages and CD4(+) T cell migration into the eye. However, inhibition of both sTNF and tmTNF is required to inhibit interferon-γ-induced chemokine receptor 2, CD40, major histocompatibility complex class II and nitric oxide (NO) up-regulation, and signalling via tmTNF is sufficient to mediate tissue damage. In confirmation, intravitreal inhibition of sTNF alone did not suppress disease, and inflammatory cells that migrated into the eye were activated, generating NO, thus causing structural damage to the retina. In contrast, intravitreal inhibition of both sTNF and tmTNF suppressed macrophage activation and therefore disease. We conclude that sTNF is required for inflammatory cell infiltration into target tissue, but at the tissue site inhibition of both sTNF and tmTNF is required to inhibit macrophage activation and to protect from tissue damage.
© 2012 The Authors;Clinical and Experimental Immunology © 2012 British Society for Immunology.
Figures
References
-
- Sedgwick JD, Riminton DS, Cyster JG, Körner H. Tumor necrosis factor: a master-regulator of leukocyte movement. Immunol Today. 2000;21:110–3. - PubMed
-
- Clark IA. How TNF was recognized as a key mechanism of disease. Cytokine Growth Factor Rev. 2007;18:335–43. - PubMed
-
- Tincani A, Andreoli L, Bazzani C, Bosiso D, Sozzani S. Inflammatory molecules: a target for treatment of systemic autoimmune diseases. Autoimmun Rev. 2007;7:1–7. - PubMed
-
- Raveney BJE, Copland DA, Dick AD, Nicholson LB. TNFR1-dependent regulation of myeloid cell function in experimental autoimmune uveoretinitis. J Immunol. 2009;183:2321–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
