

CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells
- PMID: 22472119
- PMCID: PMC3367125
- DOI: 10.1158/0008-5472.CAN-11-3567
CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells
Abstract
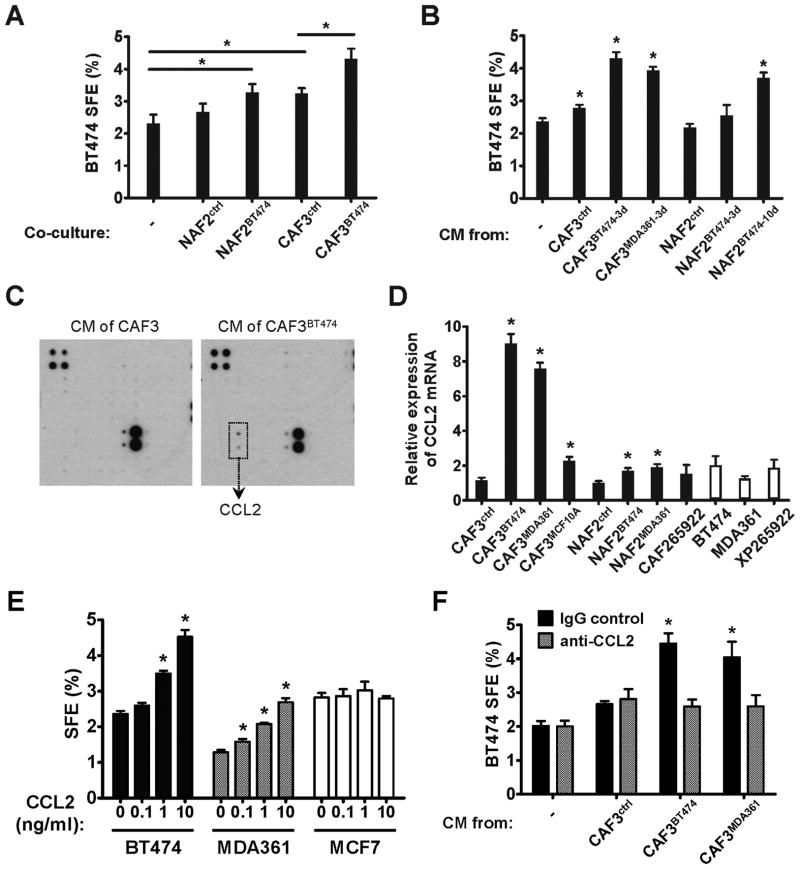
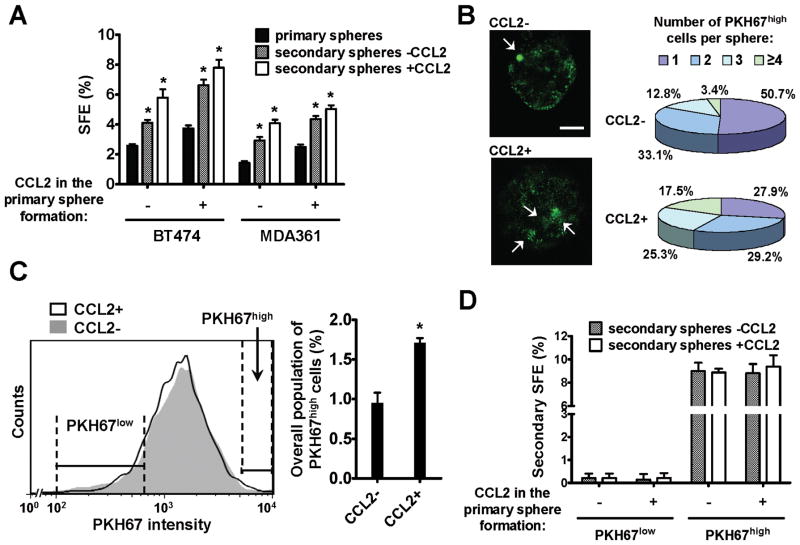
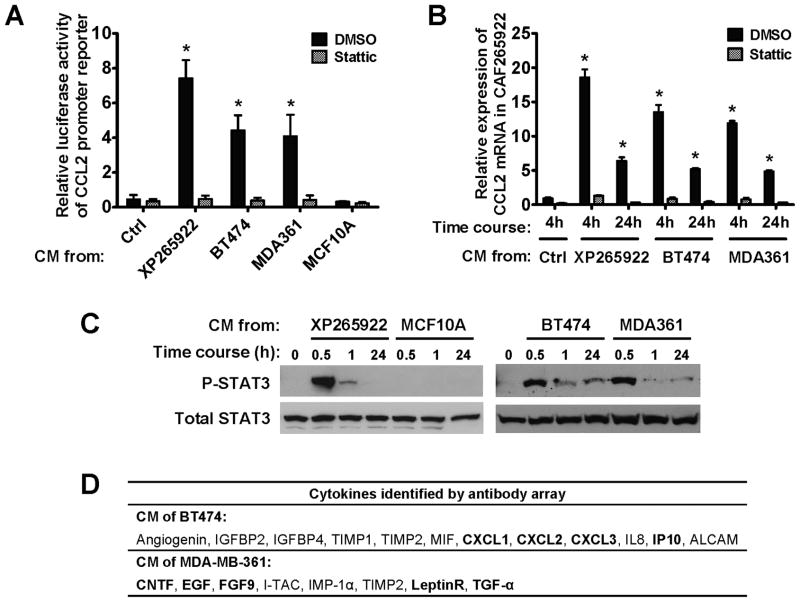
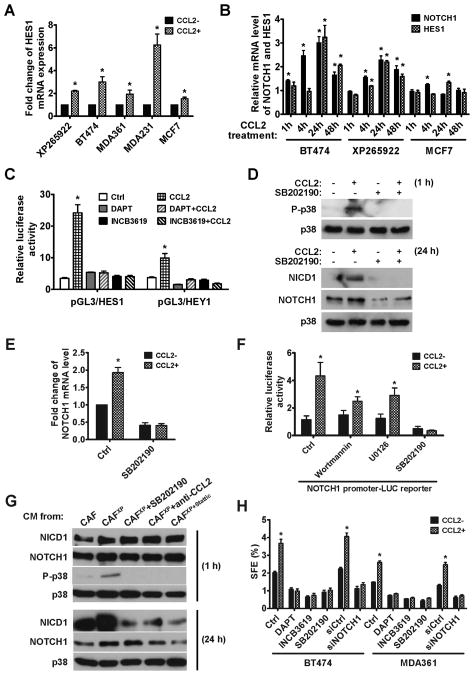
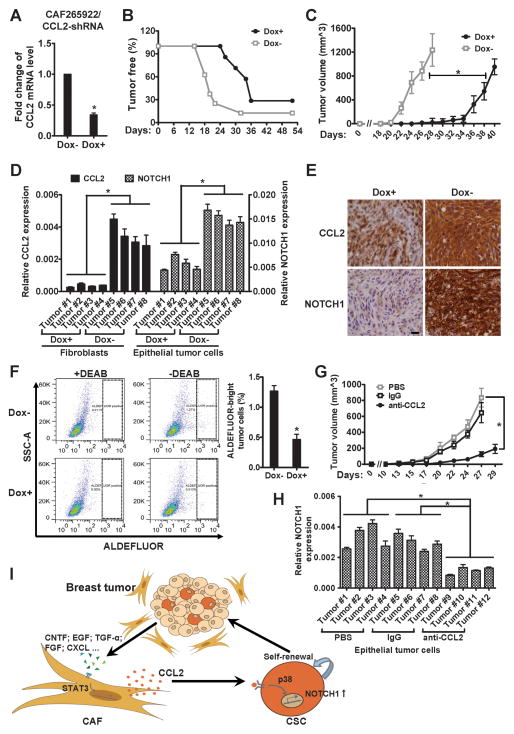
Cancer stem cells (CSC) play critical roles in cancer initiation, progression, and therapeutic refractoriness. Although many studies have focused on the genes and pathways involved in stemness, characterization of the factors in the tumor microenvironment that regulate CSCs is lacking. In this study, we investigated the effects of stromal fibroblasts on breast cancer stem cells. We found that compared with normal fibroblasts, primary cancer-associated fibroblasts (CAF) and fibroblasts activated by cocultured breast cancer cells produce higher levels of chemokine (C-C motif) ligand 2 (CCL2), which stimulates the stem cell-specific, sphere-forming phenotype in breast cancer cells and CSC self-renewal. Increased CCL2 expression in activated fibroblasts required STAT3 activation by diverse breast cancer-secreted cytokines, and in turn, induced NOTCH1 expression and the CSC features in breast cancer cells, constituting a cancer-stroma-cancer signaling circuit. In a xenograft model of paired fibroblasts and breast cancer tumor cells, loss of CCL2 significantly inhibited tumorigenesis and NOTCH1 expression. In addition, upregulation of both NOTCH1 and CCL2 was associated with poor differentiation in primary breast cancers, further supporting the observation that NOTCH1 is regulated by CCL2. Our findings therefore suggest that CCL2 represents a potential therapeutic target that can block the cancer-host communication that prompts CSC-mediated disease progression.
©2012 AACR
Conflict of interest statement
Figures
Comment in
-
Fibroblast-derived CCL2 induces cancer stem cells--response.Cancer Res. 2013 Jan 15;73(2):1032-3. doi: 10.1158/0008-5472.CAN-12-3194. Epub 2013 Jan 10. Cancer Res. 2013. PMID: 23316033 Free PMC article. No abstract available.
-
Fibroblast-derived CCL2 induces cancer stem cells--letter.Cancer Res. 2013 Jan 15;73(2):1031. doi: 10.1158/0008-5472.CAN-12-2379. Epub 2013 Jan 10. Cancer Res. 2013. PMID: 23316034 No abstract available.
References
-
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–68. - PubMed
-
- Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol. 2007;18(5):460–6. - PubMed
-
- Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. - PubMed
-
- Melgarejo E, Medina MA, Sanchez-Jimenez F, Urdiales JL. Monocyte chemoattractant protein-1: a key mediator in inflammatory processes. Int J Biochem Cell Biol. 2009;41(5):998–1001. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
