

Accurate prediction of inducible transcription factor binding intensities in vivo
- PMID: 22479205
- PMCID: PMC3315474
- DOI: 10.1371/journal.pgen.1002610
Accurate prediction of inducible transcription factor binding intensities in vivo
Abstract
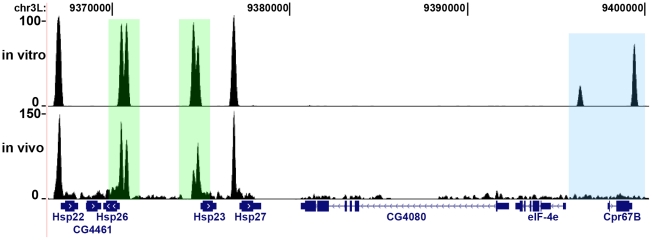
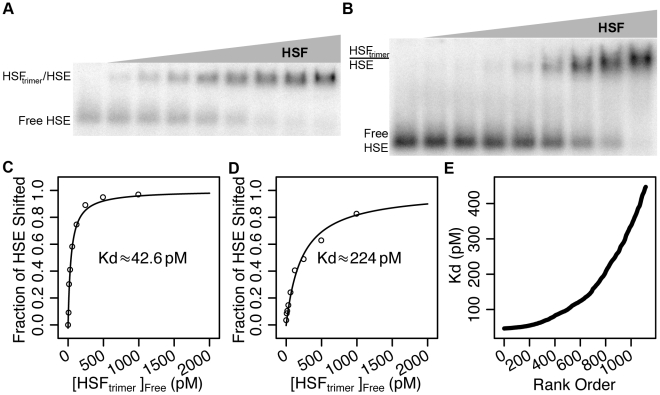
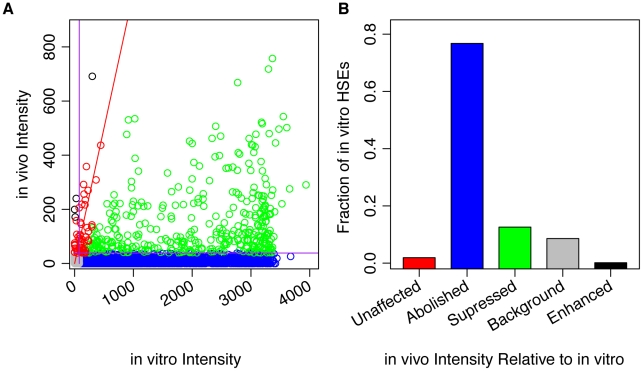
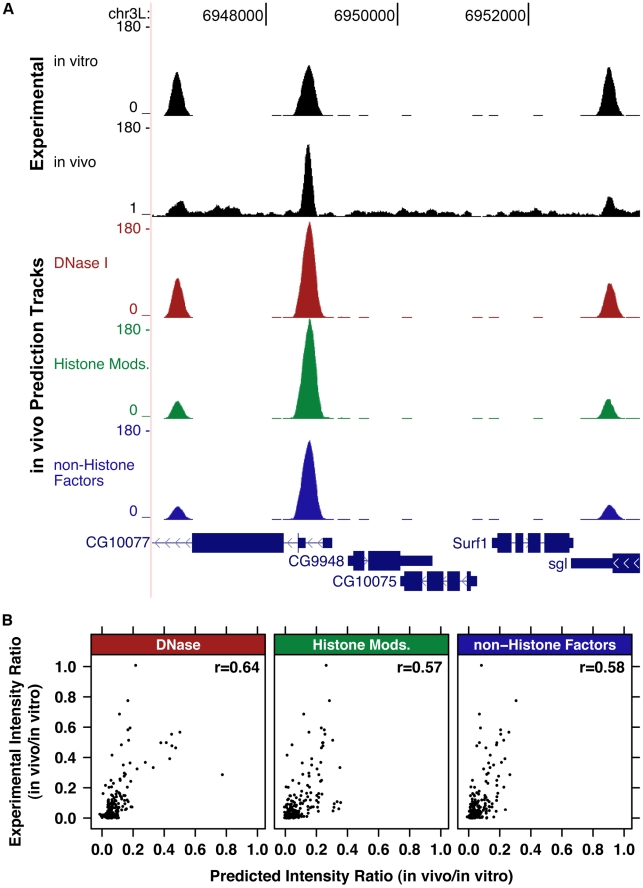
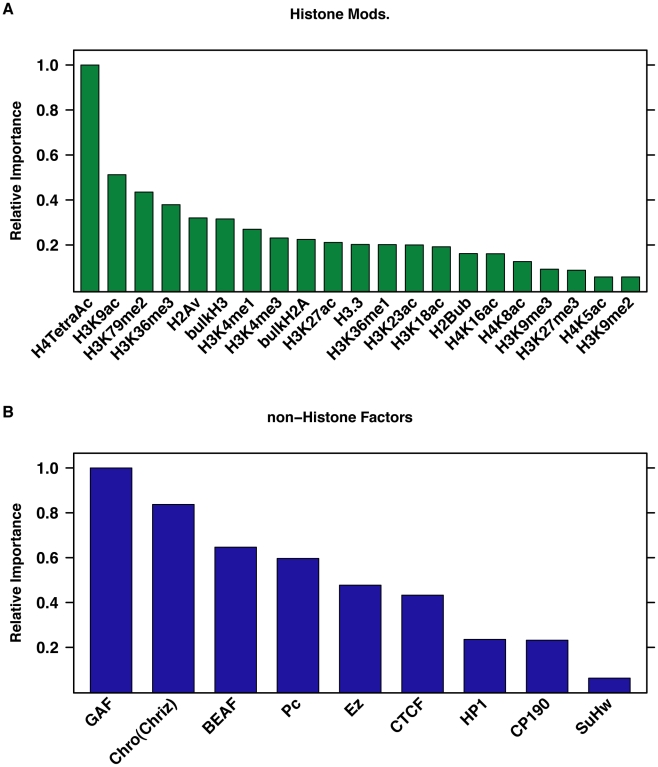
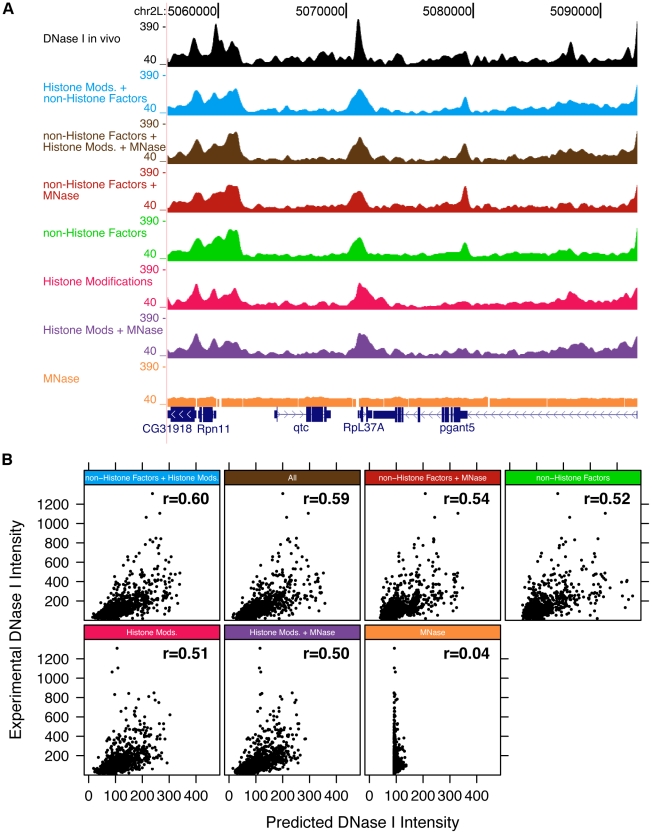
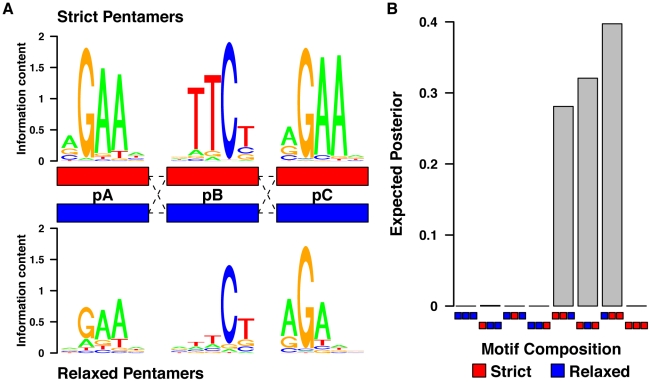
DNA sequence and local chromatin landscape act jointly to determine transcription factor (TF) binding intensity profiles. To disentangle these influences, we developed an experimental approach, called protein/DNA binding followed by high-throughput sequencing (PB-seq), that allows the binding energy landscape to be characterized genome-wide in the absence of chromatin. We applied our methods to the Drosophila Heat Shock Factor (HSF), which inducibly binds a target DNA sequence element (HSE) following heat shock stress. PB-seq involves incubating sheared naked genomic DNA with recombinant HSF, partitioning the HSF-bound and HSF-free DNA, and then detecting HSF-bound DNA by high-throughput sequencing. We compared PB-seq binding profiles with ones observed in vivo by ChIP-seq and developed statistical models to predict the observed departures from idealized binding patterns based on covariates describing the local chromatin environment. We found that DNase I hypersensitivity and tetra-acetylation of H4 were the most influential covariates in predicting changes in HSF binding affinity. We also investigated the extent to which DNA accessibility, as measured by digital DNase I footprinting data, could be predicted from MNase-seq data and the ChIP-chip profiles for many histone modifications and TFs, and found GAGA element associated factor (GAF), tetra-acetylation of H4, and H4K16 acetylation to be the most predictive covariates. Lastly, we generated an unbiased model of HSF binding sequences, which revealed distinct biophysical properties of the HSF/HSE interaction and a previously unrecognized substructure within the HSE. These findings provide new insights into the interplay between the genomic sequence and the chromatin landscape in determining transcription factor binding intensity.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Field Y, Sharon E, Segal E. How transcription factors identify regulatory sites in genomic sequence. Subcell Biochem. 2011;52:193–204. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
