

Membrane docking geometry of GRP1 PH domain bound to a target lipid bilayer: an EPR site-directed spin-labeling and relaxation study
- PMID: 22479423
- PMCID: PMC3316598
- DOI: 10.1371/journal.pone.0033640
Membrane docking geometry of GRP1 PH domain bound to a target lipid bilayer: an EPR site-directed spin-labeling and relaxation study
Abstract
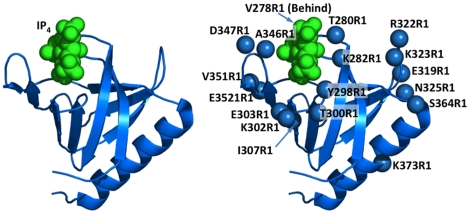
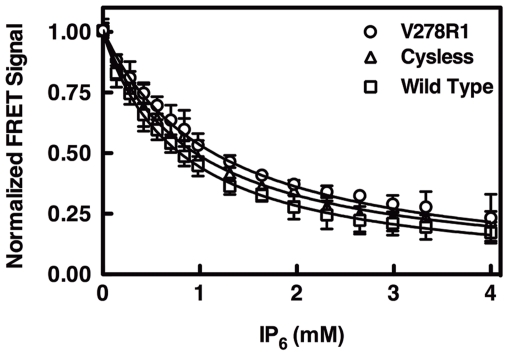
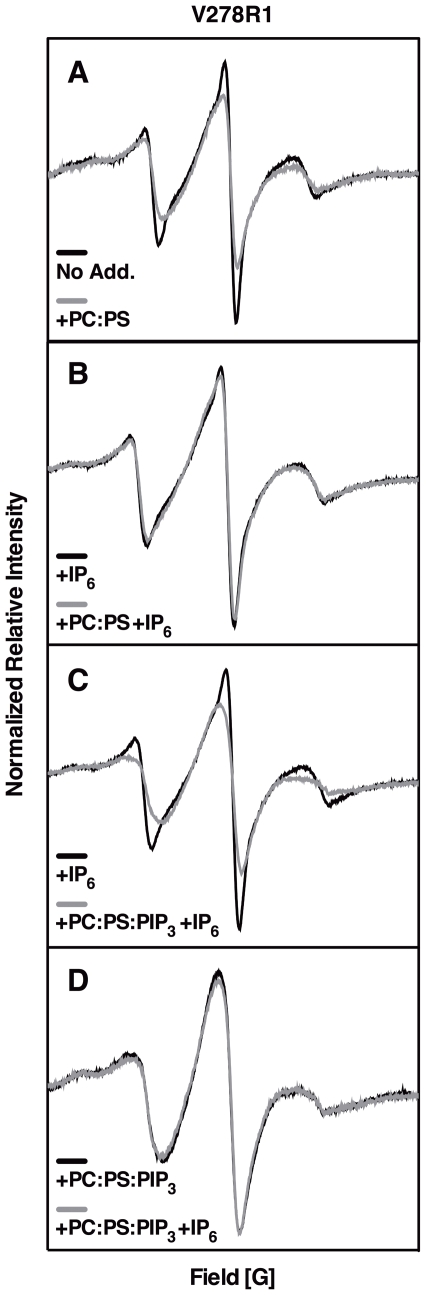
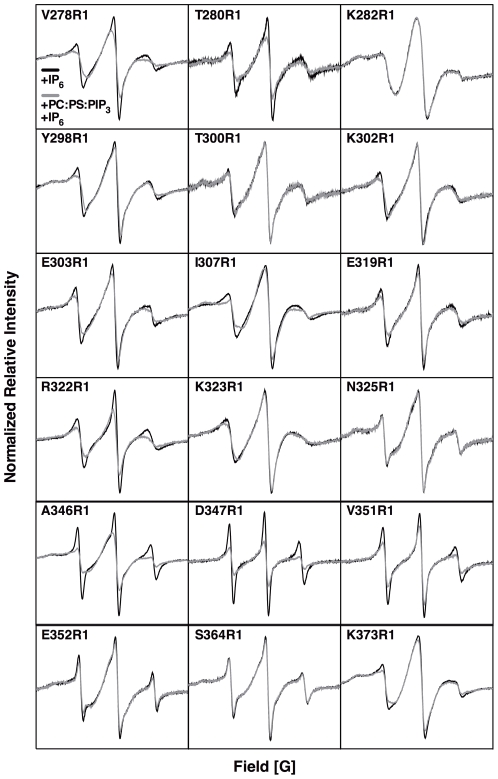
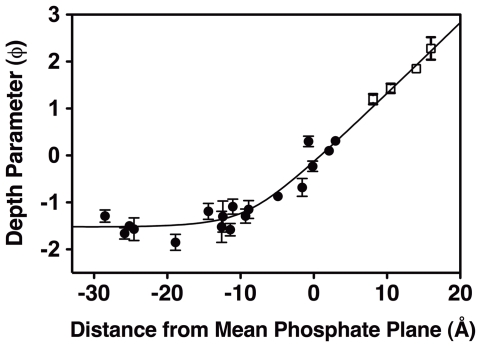
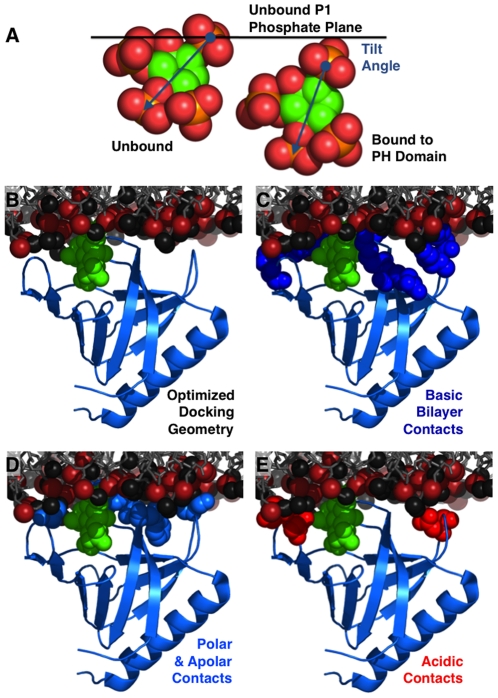
The second messenger lipid PIP(3) (phosphatidylinositol-3,4,5-trisphosphate) is generated by the lipid kinase PI3K (phosphoinositide-3-kinase) in the inner leaflet of the plasma membrane, where it regulates a broad array of cell processes by recruiting multiple signaling proteins containing PIP(3)-specific pleckstrin homology (PH) domains to the membrane surface. Despite the broad importance of PIP(3)-specific PH domains, the membrane docking geometry of a PH domain bound to its target PIP(3) lipid on a bilayer surface has not yet been experimentally determined. The present study employs EPR site-directed spin labeling and relaxation methods to elucidate the membrane docking geometry of GRP1 PH domain bound to bilayer-embedded PIP(3). The model target bilayer contains the neutral background lipid PC and both essential targeting lipids: (i) PIP(3) target lipid that provides specificity and affinity, and (ii) PS facilitator lipid that enhances the PIP(3) on-rate via an electrostatic search mechanism. The EPR approach measures membrane depth parameters for 18 function-retaining spin labels coupled to the PH domain, and for calibration spin labels coupled to phospholipids. The resulting depth parameters, together with the known high resolution structure of the co-complex between GRP1 PH domain and the PIP(3) headgroup, provide sufficient constraints to define an optimized, self-consistent membrane docking geometry. In this optimized geometry the PH domain engulfs the PIP(3) headgroup with minimal bilayer penetration, yielding the shallowest membrane position yet described for a lipid binding domain. This binding interaction displaces the PIP(3) headgroup from its lowest energy position and orientation in the bilayer, but the headgroup remains within its energetically accessible depth and angular ranges. Finally, the optimized docking geometry explains previous biophysical findings including mutations observed to disrupt membrane binding, and the rapid lateral diffusion observed for PIP(3)-bound GRP1 PH domain on supported lipid bilayers.
Conflict of interest statement
Figures
Similar articles
-
Effect of PIP2 binding on the membrane docking geometry of PKC alpha C2 domain: an EPR site-directed spin-labeling and relaxation study.Biochemistry. 2008 Aug 12;47(32):8301-16. doi: 10.1021/bi800711t. Epub 2008 Jul 9. Biochemistry. 2008. PMID: 18610985 Free PMC article.
-
Molecular mechanism of membrane binding of the GRP1 PH domain.J Mol Biol. 2013 Sep 9;425(17):3073-90. doi: 10.1016/j.jmb.2013.05.026. Epub 2013 Jun 6. J Mol Biol. 2013. PMID: 23747485 Free PMC article.
-
GRP1 pleckstrin homology domain: activation parameters and novel search mechanism for rare target lipid.Biochemistry. 2004 Dec 28;43(51):16161-73. doi: 10.1021/bi049017a. Biochemistry. 2004. PMID: 15610010 Free PMC article.
-
Does the Lipid Bilayer Orchestrate Access and Binding of Ligands to Transmembrane Orthosteric/Allosteric Sites of G Protein-Coupled Receptors?Mol Pharmacol. 2019 Nov;96(5):527-541. doi: 10.1124/mol.118.115113. Epub 2019 Apr 8. Mol Pharmacol. 2019. PMID: 30967440 Free PMC article. Review.
-
Physical properties of lipid bilayers from EPR spin labeling and their influence on chemical reactions in a membrane environment.Free Radic Biol Med. 2009 Mar 15;46(6):707-18. doi: 10.1016/j.freeradbiomed.2008.11.024. Epub 2008 Dec 11. Free Radic Biol Med. 2009. PMID: 19111611 Free PMC article. Review.
Cited by
-
Counterion-mediated cluster formation by polyphosphoinositides.Chem Phys Lipids. 2014 Sep;182:38-51. doi: 10.1016/j.chemphyslip.2014.01.001. Epub 2014 Jan 15. Chem Phys Lipids. 2014. PMID: 24440472 Free PMC article. Review.
-
Solvent-Free, Highly Coarse-Grained Models for Charged Lipid Systems.J Chem Theory Comput. 2014 Oct 14;10(10):4730-4744. doi: 10.1021/ct500474a. Epub 2014 Sep 10. J Chem Theory Comput. 2014. PMID: 25328498 Free PMC article.
-
Membrane Docking of the Synaptotagmin 7 C2A Domain: Electron Paramagnetic Resonance Measurements Show Contributions from Two Membrane Binding Loops.Biochemistry. 2015 Sep 22;54(37):5684-95. doi: 10.1021/acs.biochem.5b00421. Epub 2015 Sep 10. Biochemistry. 2015. PMID: 26322740 Free PMC article.
-
Membrane surface recognition by the ASAP1 PH domain and consequences for interactions with the small GTPase Arf1.Sci Adv. 2020 Sep 30;6(40):eabd1882. doi: 10.1126/sciadv.abd1882. Print 2020 Sep. Sci Adv. 2020. PMID: 32998886 Free PMC article.
-
Multiple lipid binding sites determine the affinity of PH domains for phosphoinositide-containing membranes.Sci Adv. 2020 Feb 19;6(8):eaay5736. doi: 10.1126/sciadv.aay5736. eCollection 2020 Feb. Sci Adv. 2020. PMID: 32128410 Free PMC article.
References
-
- Czech MP. PIP2 and PIP3: complex roles at the cell surface. Cell. 2000;100:603–606. - PubMed
-
- Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. - PubMed
-
- Wang F, Herzmark P, Weiner OD, Srinivasan S, Servant G, et al. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat Cell Biol. 2002;4:513–518. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
