

Network analysis of epidermal growth factor signaling using integrated genomic, proteomic and phosphorylation data
- PMID: 22479638
- PMCID: PMC3315547
- DOI: 10.1371/journal.pone.0034515
Network analysis of epidermal growth factor signaling using integrated genomic, proteomic and phosphorylation data
Abstract
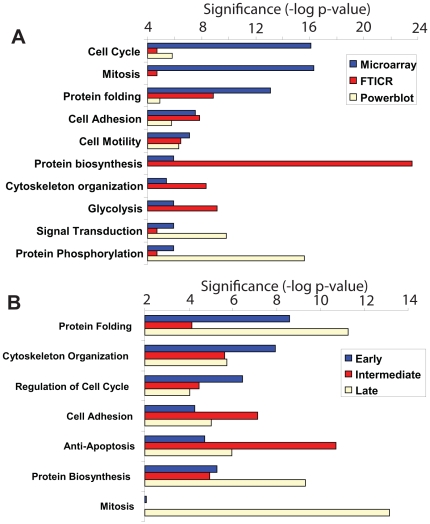
To understand how integration of multiple data types can help decipher cellular responses at the systems level, we analyzed the mitogenic response of human mammary epithelial cells to epidermal growth factor (EGF) using whole genome microarrays, mass spectrometry-based proteomics and large-scale western blots with over 1000 antibodies. A time course analysis revealed significant differences in the expression of 3172 genes and 596 proteins, including protein phosphorylation changes measured by western blot. Integration of these disparate data types showed that each contributed qualitatively different components to the observed cell response to EGF and that varying degrees of concordance in gene expression and protein abundance measurements could be linked to specific biological processes. Networks inferred from individual data types were relatively limited, whereas networks derived from the integrated data recapitulated the known major cellular responses to EGF and exhibited more highly connected signaling nodes than networks derived from any individual dataset. While cell cycle regulatory pathways were altered as anticipated, we found the most robust response to mitogenic concentrations of EGF was induction of matrix metalloprotease cascades, highlighting the importance of the EGFR system as a regulator of the extracellular environment. These results demonstrate the value of integrating multiple levels of biological information to more accurately reconstruct networks of cellular response.
Conflict of interest statement
Figures





References
-
- Aderem A. Systems biology: its practice and challenges. Cell. 2005;121:511–513. - PubMed
-
- Wiley HS, Shvartsman SY, Lauffenburger DA. Computational modeling of the EGF-receptor system: a paradigm for systems biology. Trends Cell Biol. 2003;13:43–50. - PubMed
-
- Nicholson JK, Holmes E, Lindon JC, Wilson ID. The challenges of modeling mammalian biocomplexity. Nat Biotechnol. 2004;22:1268–1274. - PubMed
-
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. - PubMed
-
- Hoheisel JD. Microarray technology: beyond transcript profiling and genotype analysis. Nat Rev Genet. 2006;7:200–210. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
