

The genomic basis of adaptive evolution in threespine sticklebacks
- PMID: 22481358
- PMCID: PMC3322419
- DOI: 10.1038/nature10944
The genomic basis of adaptive evolution in threespine sticklebacks
Abstract
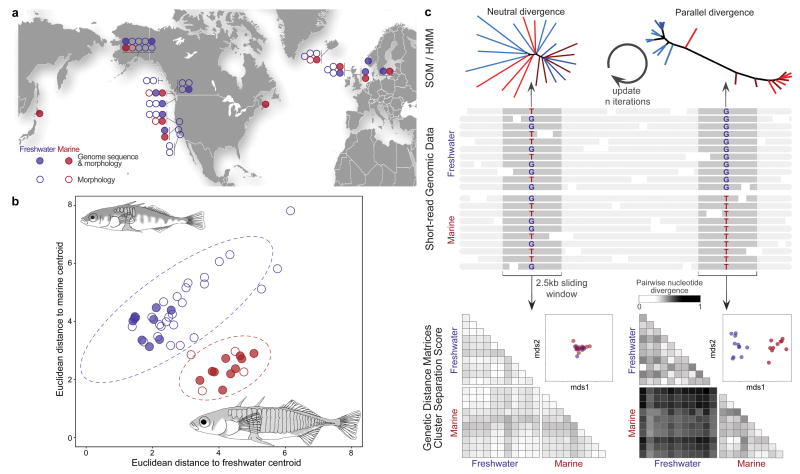
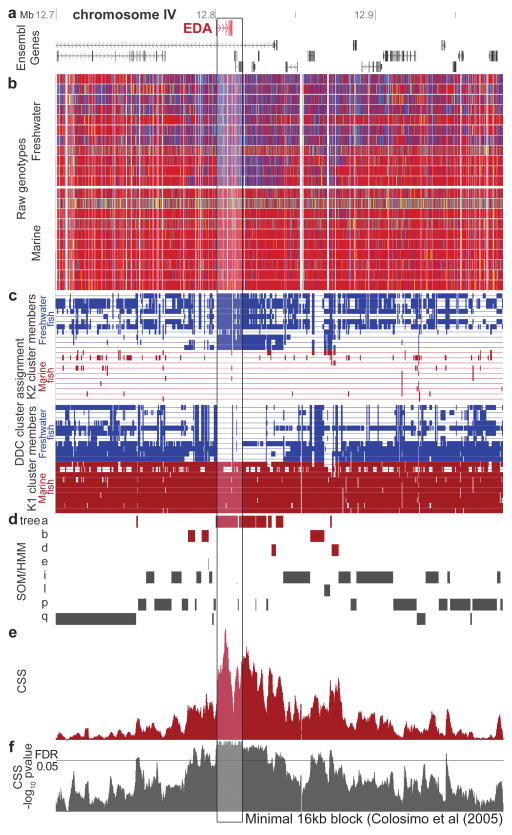
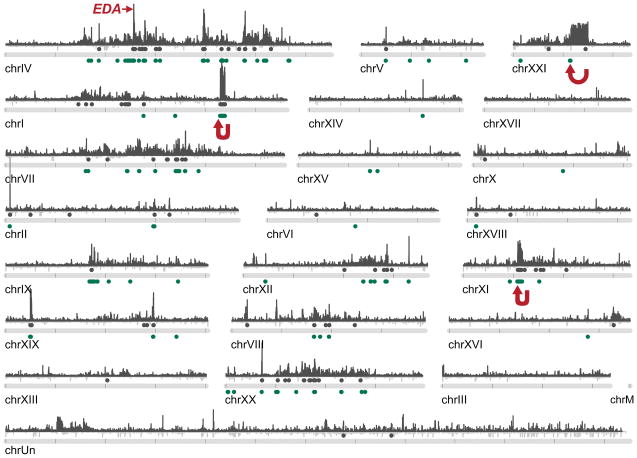
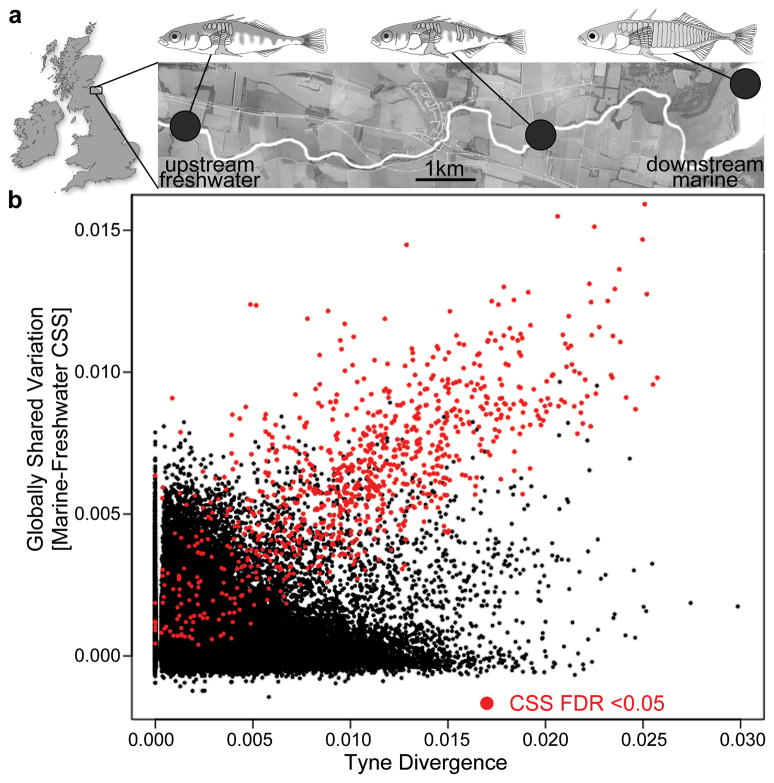
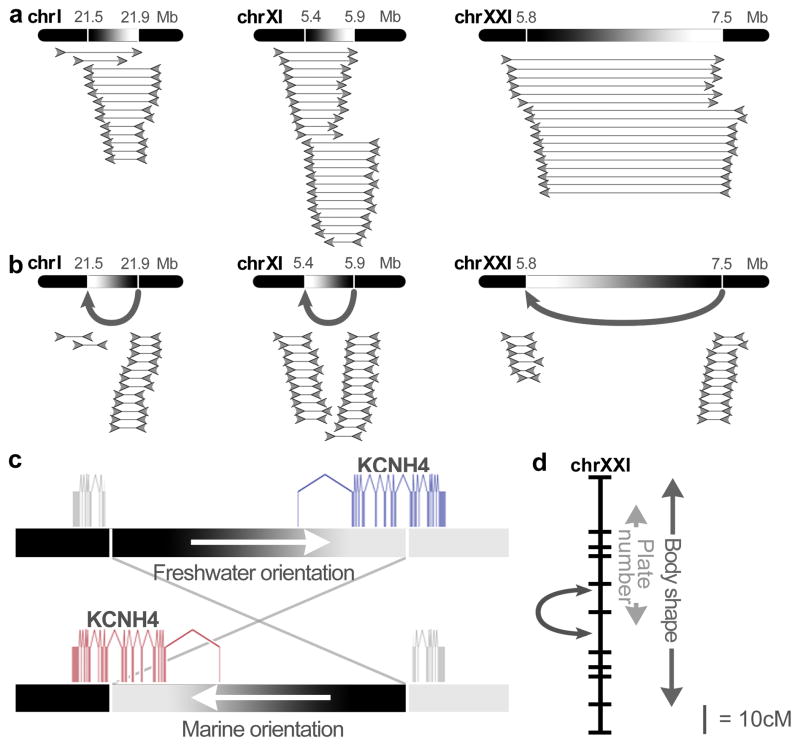
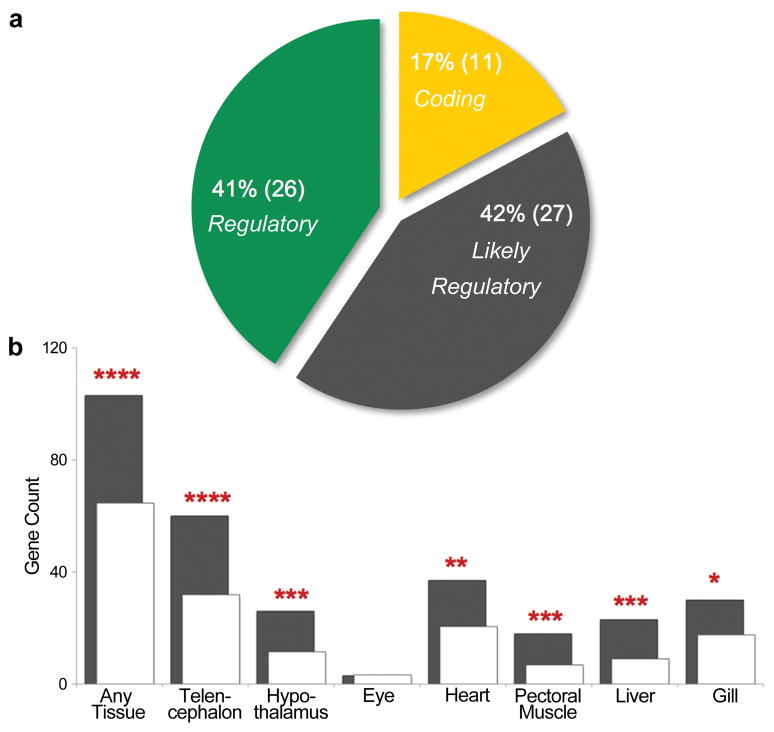
Marine stickleback fish have colonized and adapted to thousands of streams and lakes formed since the last ice age, providing an exceptional opportunity to characterize genomic mechanisms underlying repeated ecological adaptation in nature. Here we develop a high-quality reference genome assembly for threespine sticklebacks. By sequencing the genomes of twenty additional individuals from a global set of marine and freshwater populations, we identify a genome-wide set of loci that are consistently associated with marine-freshwater divergence. Our results indicate that reuse of globally shared standing genetic variation, including chromosomal inversions, has an important role in repeated evolution of distinct marine and freshwater sticklebacks, and in the maintenance of divergent ecotypes during early stages of reproductive isolation. Both coding and regulatory changes occur in the set of loci underlying marine-freshwater evolution, but regulatory changes appear to predominate in this well known example of repeated adaptive evolution in nature.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Genomics: Stickleback is the catch of the day.Nature. 2012 Apr 4;484(7392):46-7. doi: 10.1038/484046a. Nature. 2012. PMID: 22481355 No abstract available.
References
-
- Stern DL. Perspective: Evolutionary developmental biology and the problem of variation. Evolution. 2000;54:1079–1091. - PubMed
-
- Carroll SB. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell. 2008;134:25–36. - PubMed
-
- Wray G. The evolutionary significance of cis-regulatory mutations. @Nat Rev Genet. 2007;8:206–216. - PubMed
-
- Hoekstra HE, Coyne JA. The locus of evolution: evo devo and the genetics of adaptation. Evolution. 2007;61:995–1016. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
