

Cystic fibrosis: a mucosal immunodeficiency syndrome
- PMID: 22481418
- PMCID: PMC3577071
- DOI: 10.1038/nm.2715
Cystic fibrosis: a mucosal immunodeficiency syndrome
Abstract
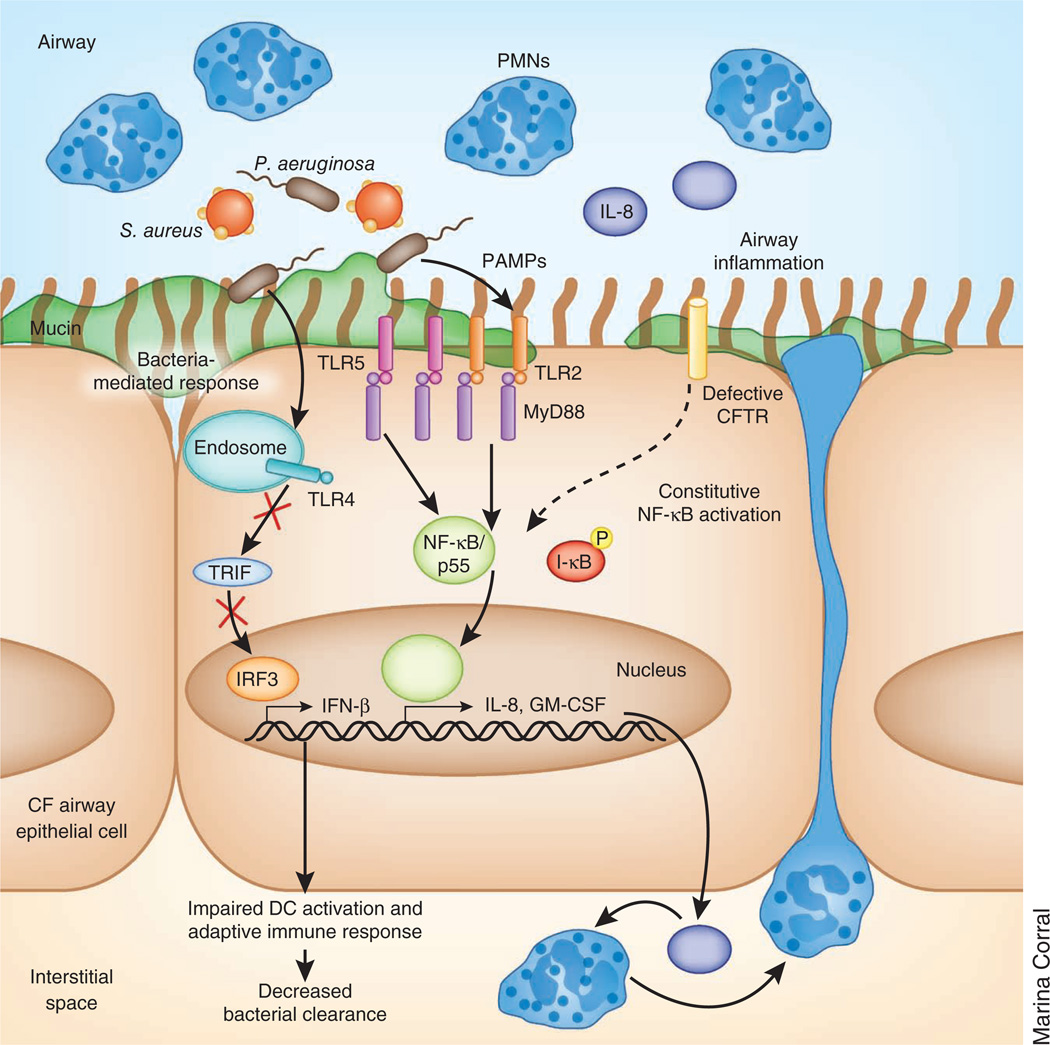
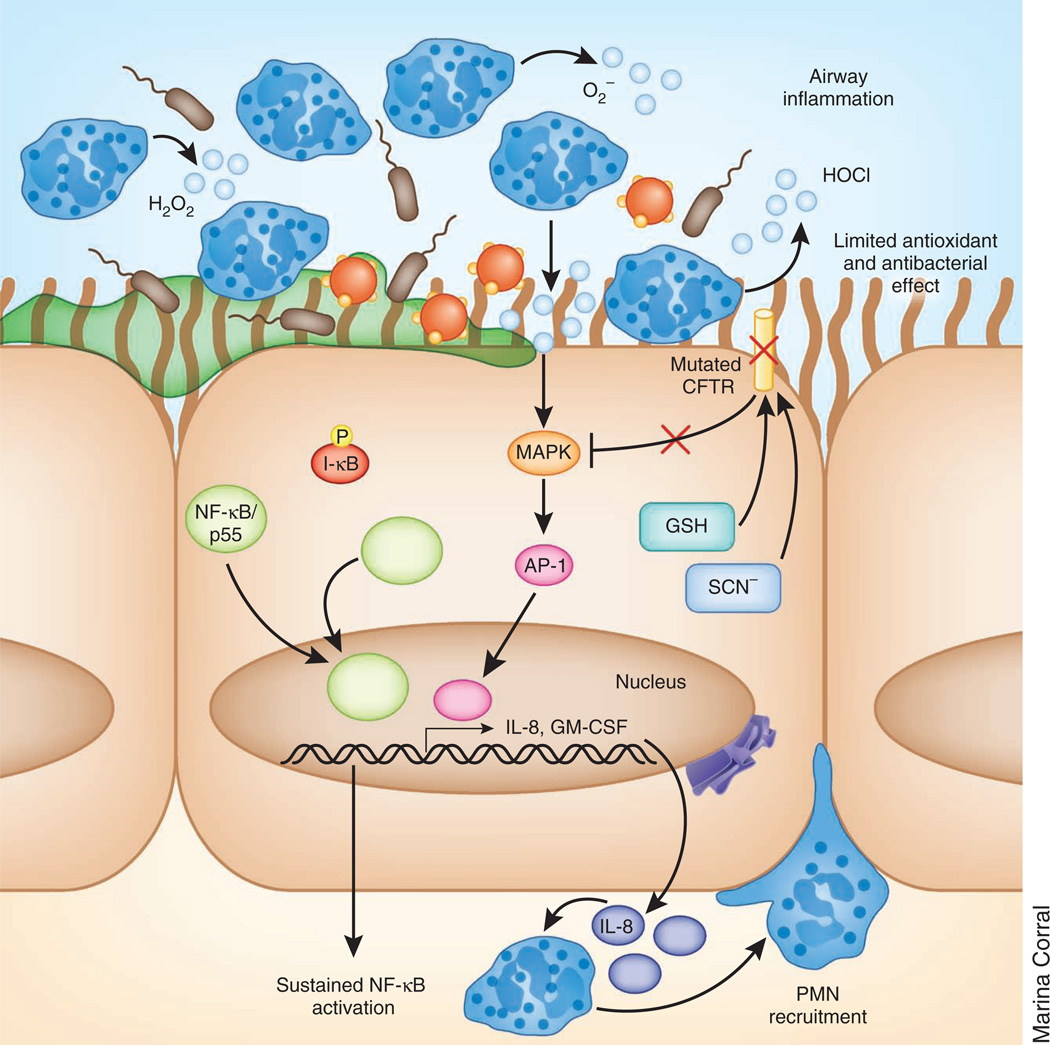
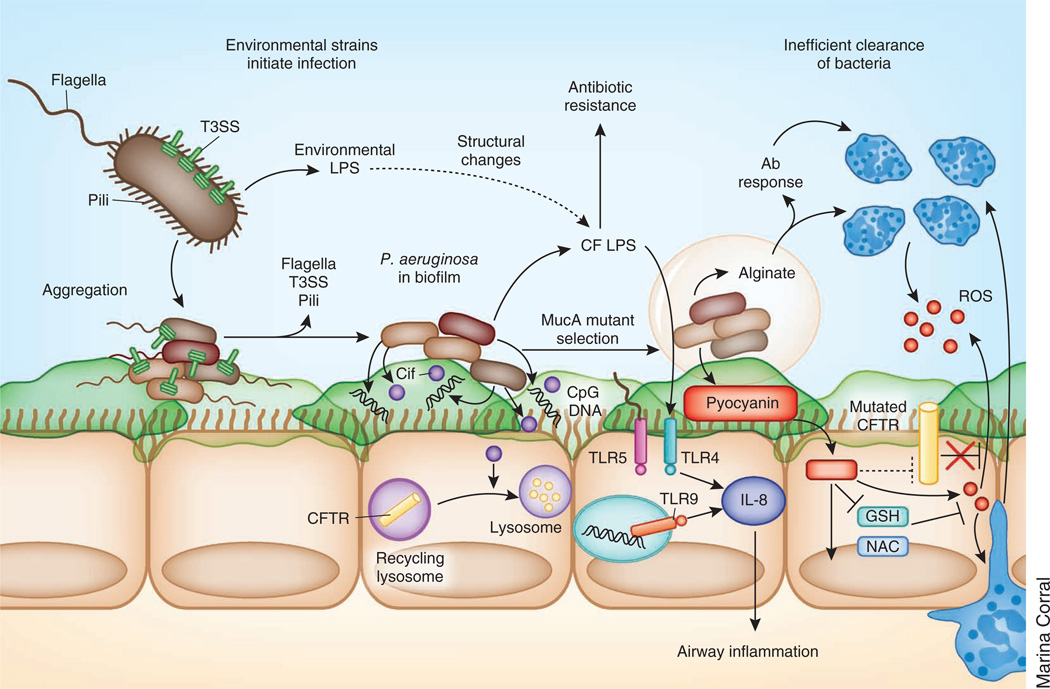
Cystic fibrosis transmembrane conductance regulator (CFTR) functions as a channel that regulates the transport of ions and the movement of water across the epithelial barrier. Mutations in CFTR, which form the basis for the clinical manifestations of cystic fibrosis, affect the epithelial innate immune function in the lung, resulting in exaggerated and ineffective airway inflammation that fails to eradicate pulmonary pathogens. Compounding the effects of excessive neutrophil recruitment, the mutant CFTR channel does not transport antioxidants to counteract neutrophil-associated oxidative stress. Whereas mutant CFTR expression in leukocytes outside of the lung does not markedly impair their function, the expected regulation of inflammation in the airways is clearly deficient in cystic fibrosis. The resulting bacterial infections, which are caused by organisms that have substantial genetic and metabolic flexibility, can resist multiple classes of antibiotics and evade phagocytic clearance. The development of animal models that approximate the human pulmonary phenotypes-airway inflammation and spontaneous infection-may provide the much-needed tools to establish how CFTR regulates mucosal immunity and to test directly the effect of pharmacologic potentiation and correction of mutant CFTR function on bacterial clearance.
Figures
References
-
- Gugler E, Pallavicini JC, Swedlow H, Zipkin I, Agnese PA. Immunological studies of submaxillary saliva from patients with cystic fibrosis and from normal children. J. Pediatr. 1968;73:548–559. - PubMed
-
- Matsui H, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95:1005–1015. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
