

Integrated inference and analysis of regulatory networks from multi-level measurements
- PMID: 22482944
- PMCID: PMC5615108
- DOI: 10.1016/B978-0-12-388403-9.00002-3
Integrated inference and analysis of regulatory networks from multi-level measurements
Abstract
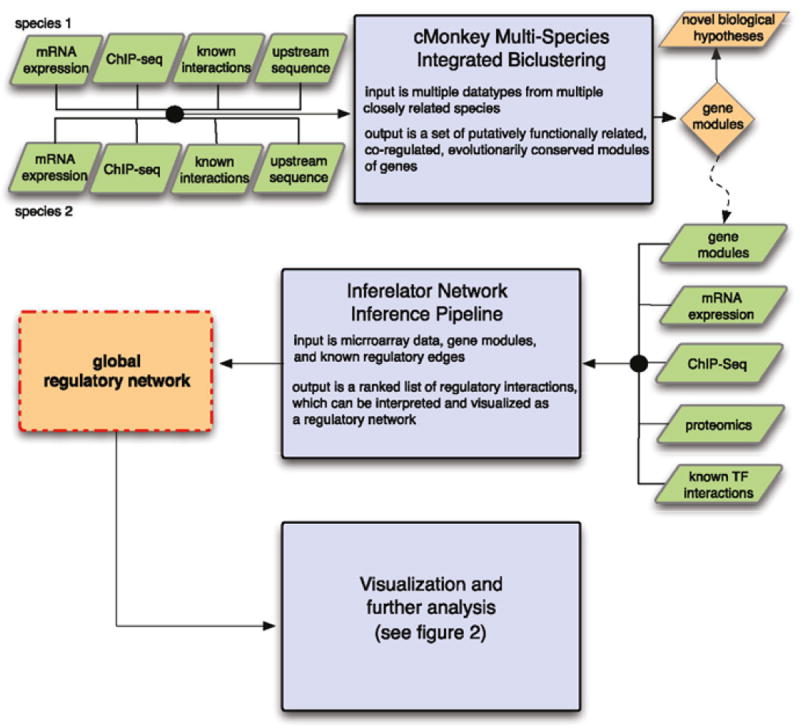
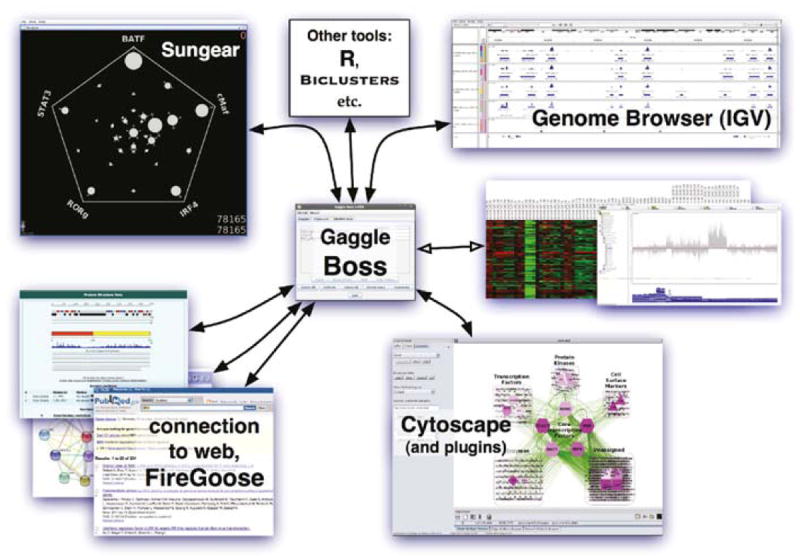
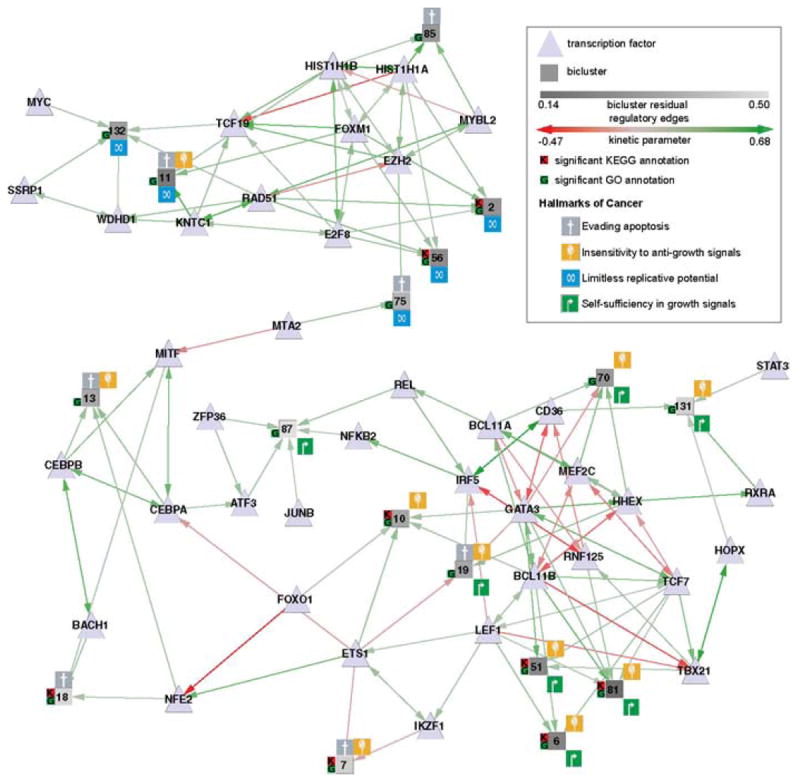
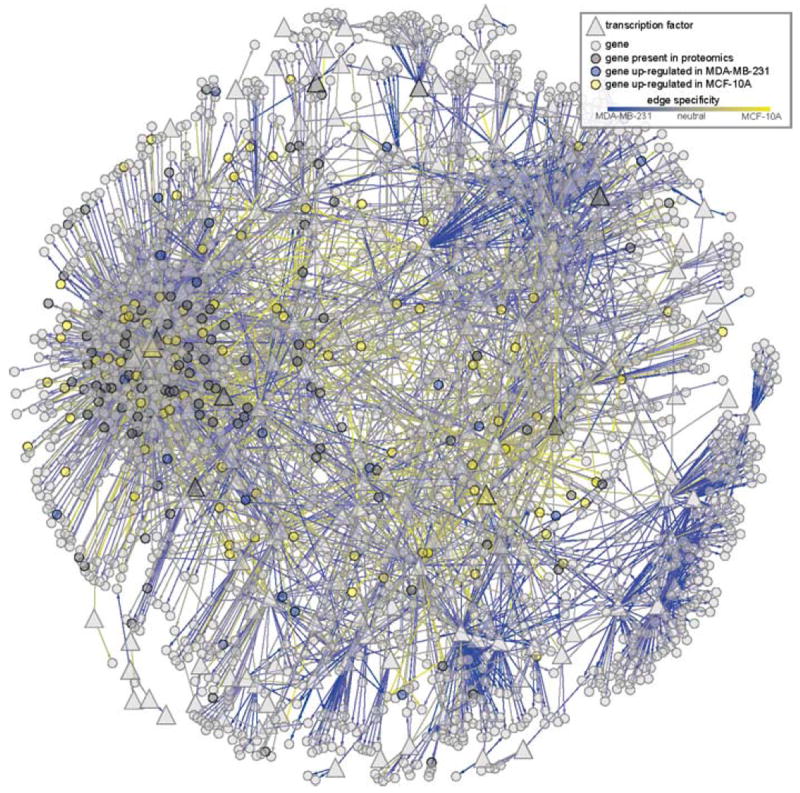
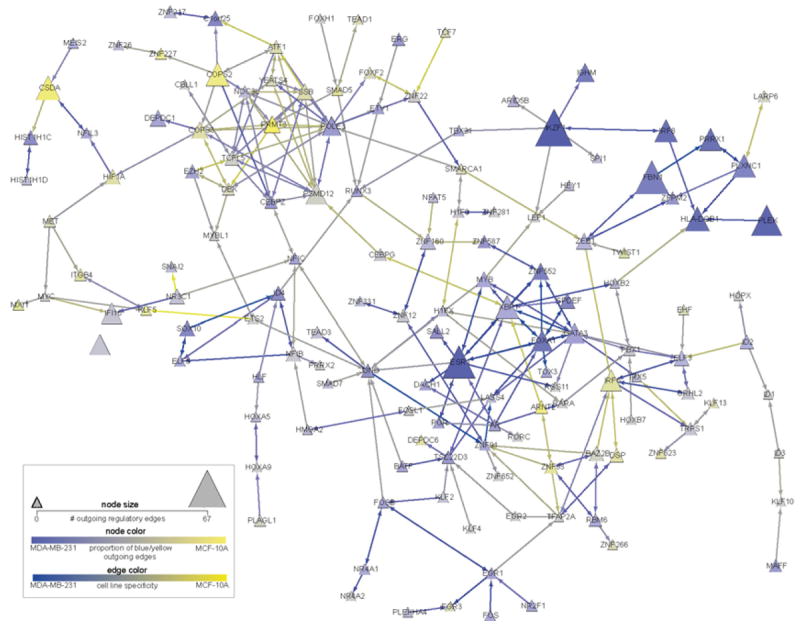
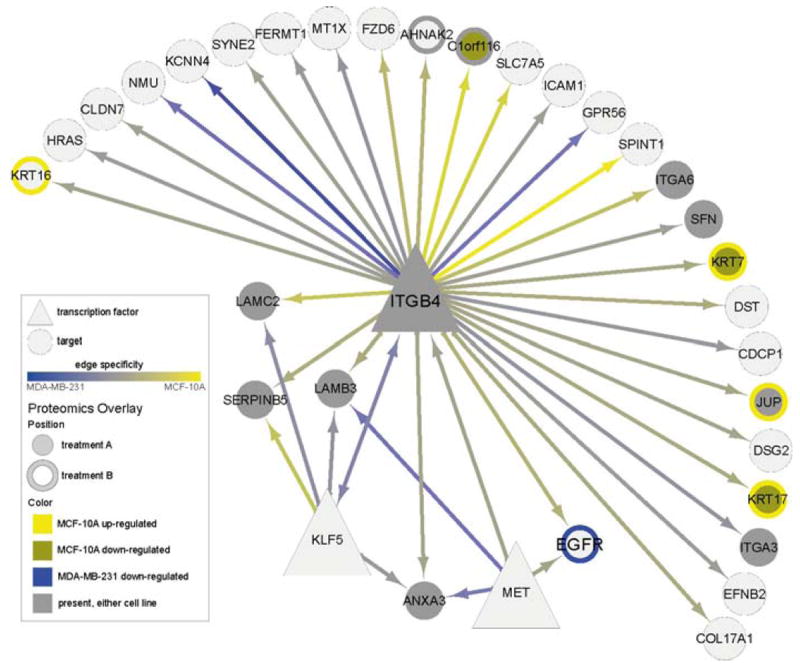
Regulatory and signaling networks coordinate the enormously complex interactions and processes that control cellular processes (such as metabolism and cell division), coordinate response to the environment, and carry out multiple cell decisions (such as development and quorum sensing). Regulatory network inference is the process of inferring these networks, traditionally from microarray data but increasingly incorporating other measurement types such as proteomics, ChIP-seq, metabolomics, and mass cytometry. We discuss existing techniques for network inference. We review in detail our pipeline, which consists of an initial biclustering step, designed to estimate co-regulated groups; a network inference step, designed to select and parameterize likely regulatory models for the control of the co-regulated groups from the biclustering step; and a visualization and analysis step, designed to find and communicate key features of the network. Learning biological networks from even the most complete data sets is challenging; we argue that integrating new data types into the inference pipeline produces networks of increased accuracy, validity, and biological relevance.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, Derow C, Feuermann M, Ghanbarian AT, Kerrien S, Khadake J, Kerssemakers J, Leroy C, Menden M, Michaut M, Montecchi-Palazzi L, Neuhauser SN, Orchard S, Perreau V, Roechert B, van Eijk K, Hermjakob H. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010;38:D525–D530. - PMC - PubMed
-
- Ashburner M, Ball CA, Blake J, ABotstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. - PMC - PubMed
-
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Int Conf Intell Syst Mol Biol; Proceedings /.. International Conference on Intelligent Systems for Molecular Biology; ISMB; 1994. pp. 28–36. - PubMed
-
- Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
