

ATP binding controls distinct structural transitions of Escherichia coli DNA gyrase in complex with DNA
- PMID: 22484318
- PMCID: PMC5660678
- DOI: 10.1038/nsmb.2278
ATP binding controls distinct structural transitions of Escherichia coli DNA gyrase in complex with DNA
Abstract
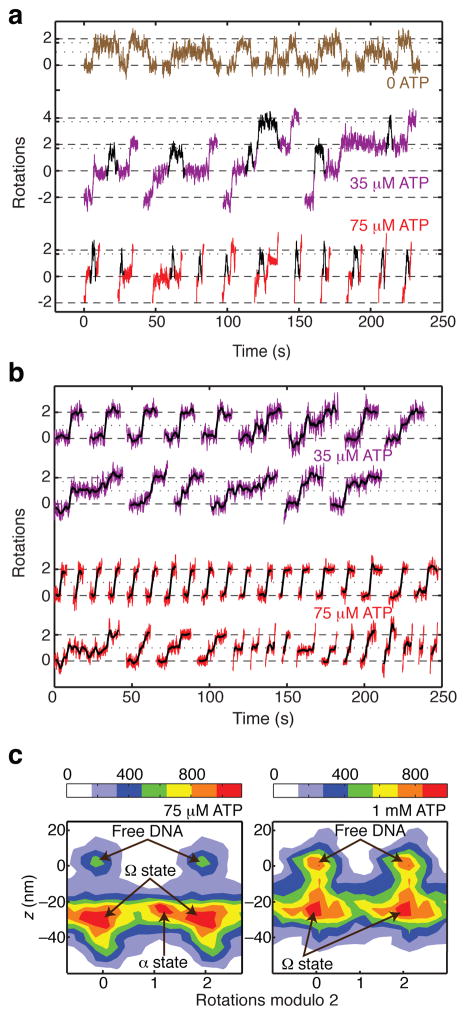
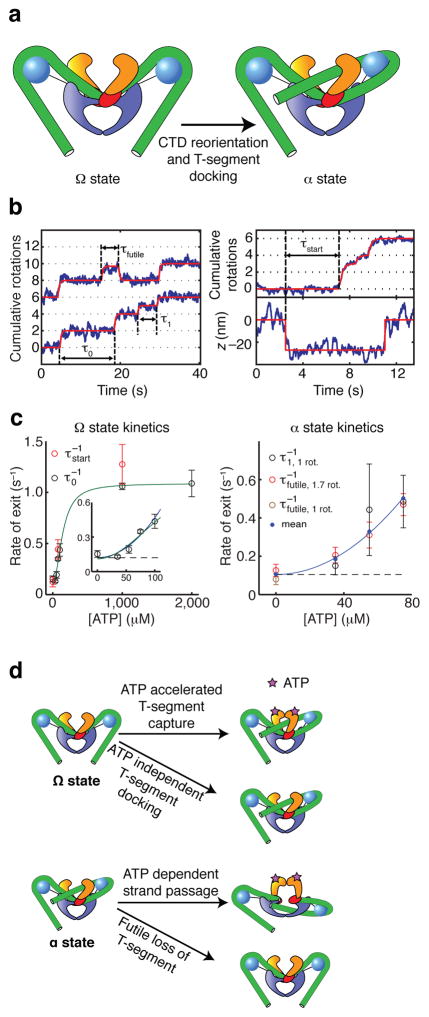
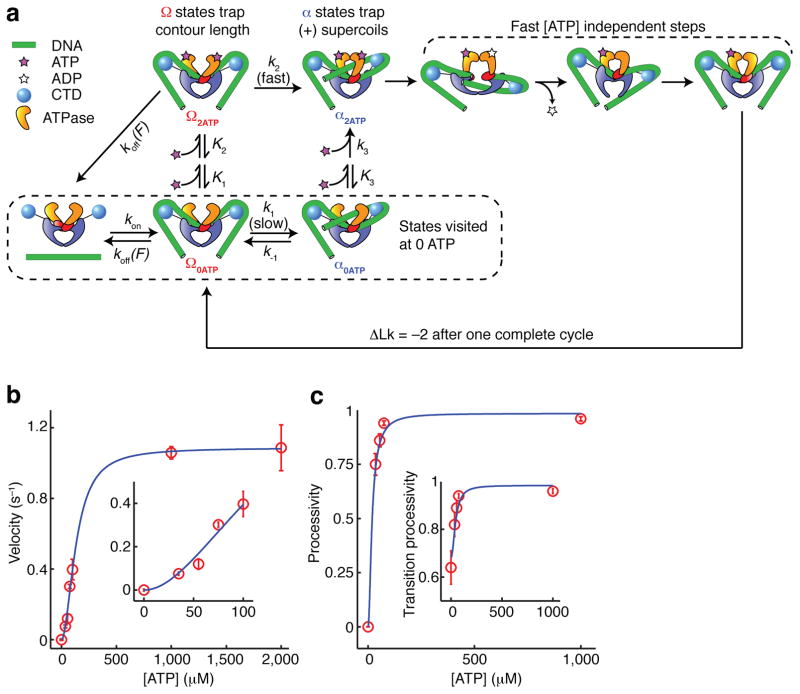
DNA gyrase is a molecular motor that harnesses the free energy of ATP hydrolysis to introduce negative supercoils into DNA. A critical step in this reaction is the formation of a chiral DNA wrap. Here we observe gyrase structural dynamics using a single-molecule assay in which gyrase drives the processive, stepwise rotation of a nanosphere attached to the side of a stretched DNA molecule. Analysis of rotational pauses and measurements of DNA contraction reveal multiple ATP-modulated structural transitions. DNA wrapping is coordinated with the ATPase cycle and proceeds by way of an unanticipated structural intermediate that dominates the kinetics of supercoiling. Our findings reveal a conformational landscape of loosely coupled transitions funneling the motor toward productive energy transduction, a feature that may be common to the reaction cycles of other DNA and protein remodeling machines.
Figures
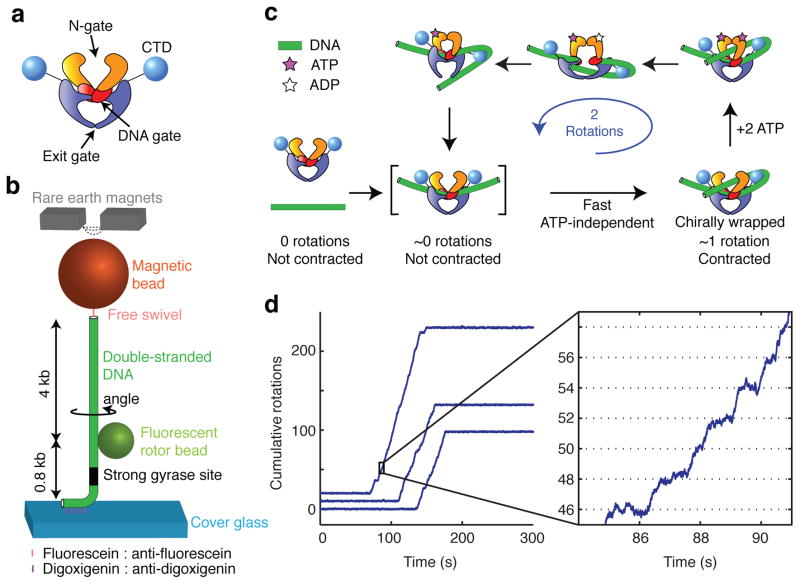
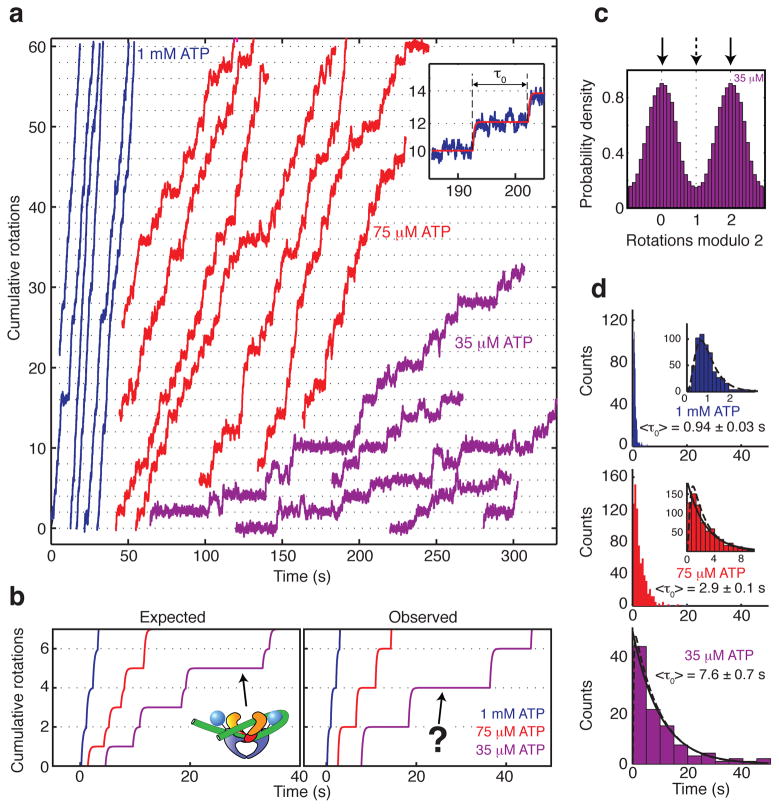
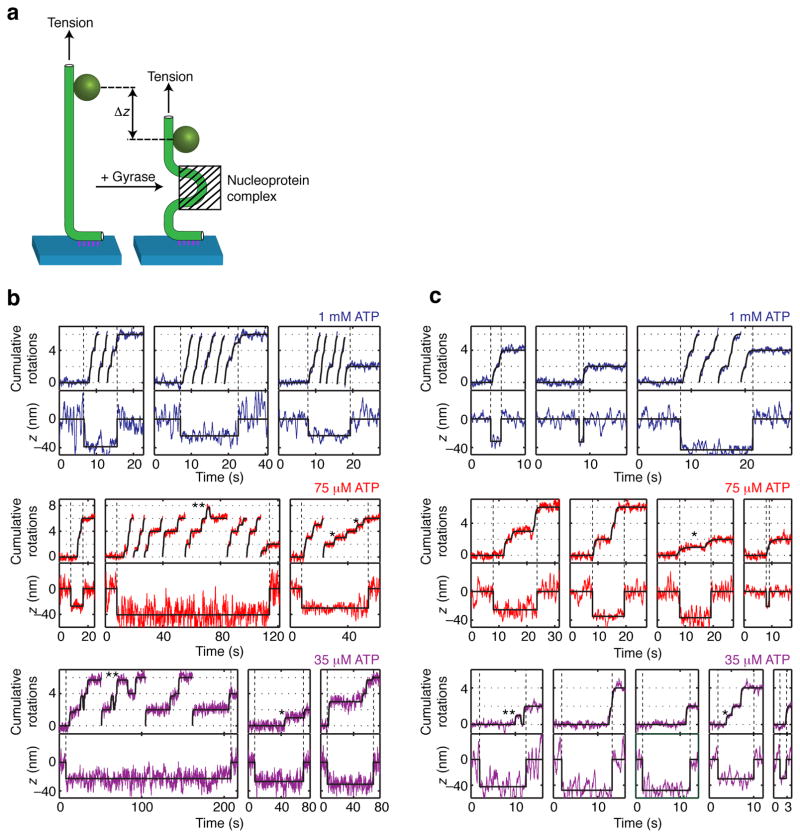
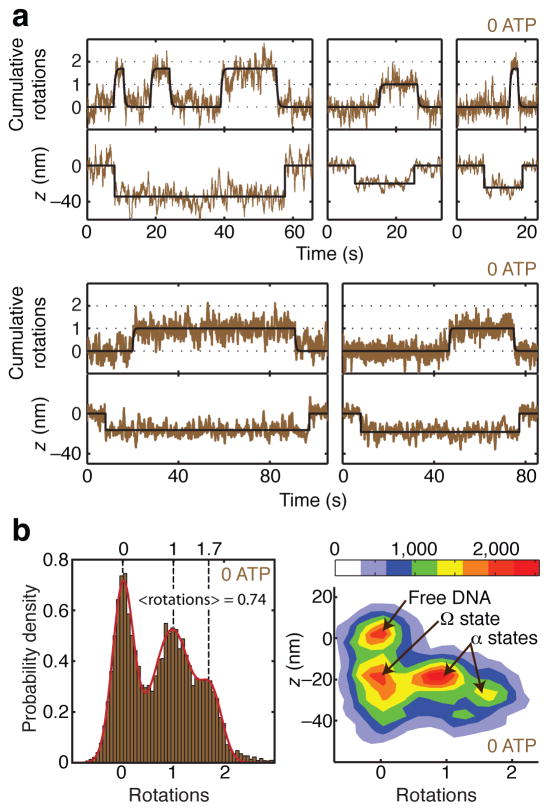
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
