

Splice site, frameshift, and chimeric GFAP mutations in Alexander disease
- PMID: 22488673
- PMCID: PMC3674965
- DOI: 10.1002/humu.22094
Splice site, frameshift, and chimeric GFAP mutations in Alexander disease
Abstract
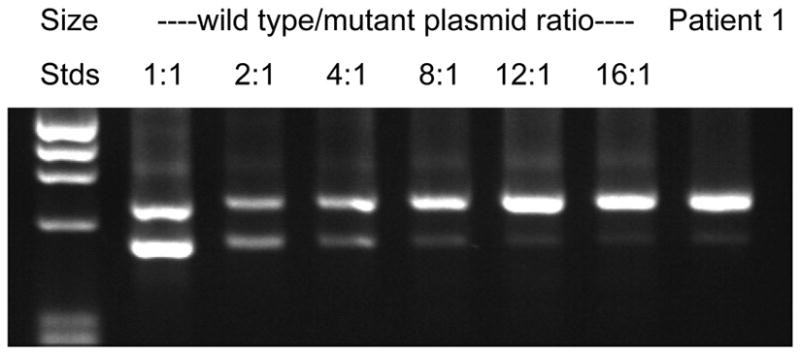
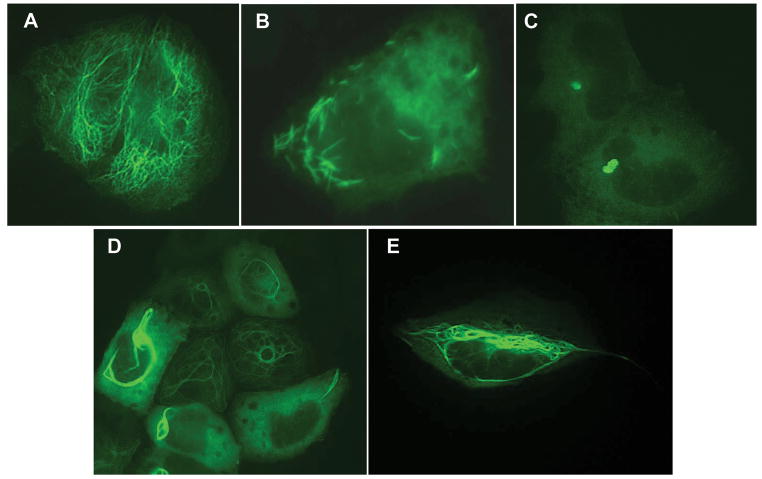
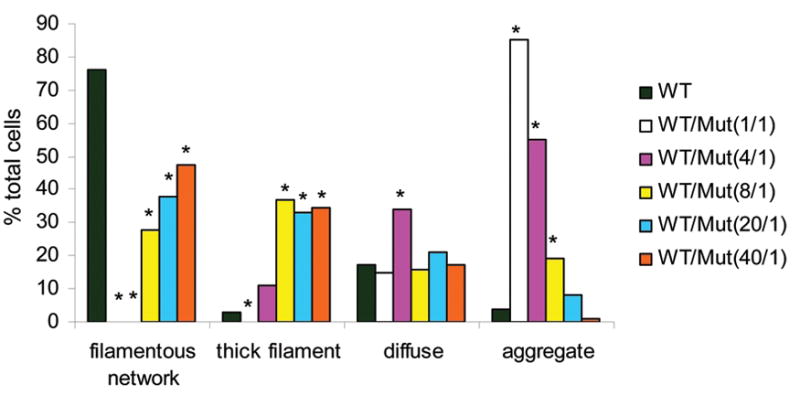
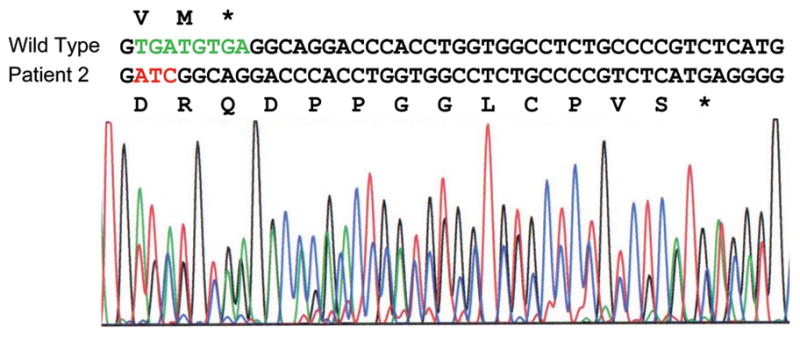
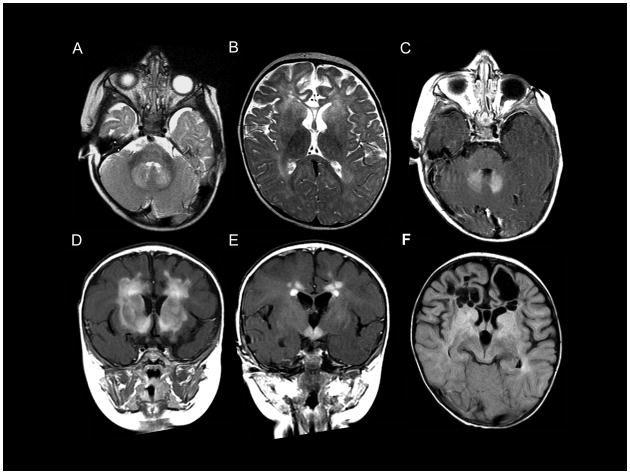
Alexander disease (AxD) is a usually fatal astrogliopathy primarily caused by mutations in the gene encoding glial fibrillary acidic protein (GFAP), an intermediate filament protein expressed in astrocytes. We describe three patients with unique characteristics, and whose mutations have implications for AxD diagnosis and studies of intermediate filaments. Patient 1 is the first reported case with a noncoding mutation. The patient has a splice site change producing an in-frame deletion of exon 4 in about 10% of the transcripts. Patient 2 has an insertion and deletion at the extreme end of the coding region, resulting in a short frameshift. In addition, the mutation was found in buccal DNA but not in blood DNA, making this patient the first reported chimera. Patient 3 has a single-base deletion near the C-terminal end of the protein, producing a short frameshift. These findings recommend inclusion of intronic splice site regions in genetic testing for AxD, indicate that alteration of only a small fraction of GFAP can produce disease, and provide caution against tagging intermediate filaments at their C-terminal end for cell biological investigations.
© 2012 Wiley Periodicals, Inc.
Figures

References
-
- Brenner M, Goldman JE, Quinlan RA, Messing A. Alexander disease: a genetic disorder of astrocytes. In: Parpura V, Haydon P, editors. Astrocytes in (patho)physiology of the nervous system. New York: Springer; 2009. pp. 591–648.
-
- Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nature Genetics. 2001;27:117–120. - PubMed
-
- Cary RB, Klymkowsky MW. Disruption of intermediate filament organization leads to structural defects at the intersomite junction in Xenopus myotomal muscle. Development. 1995;121:1041–1052. - PubMed
-
- Chin SS, Macioce P, Liem RK. Effects of truncated neurofilament proteins on the endogenous intermediate filaments in transfected fibroblasts. J Cell Sci. 1991;99 (Pt 2):335–350. - PubMed
-
- Christian JL, Edelstein NG, Moon RT. Overexpression of wild-type and dominant negative mutant vimentin subunits in developing Xenopus embryos. New Biol. 1990;2:700–711. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
