

Cross-suppression of EGFR ligands amphiregulin and epiregulin and de-repression of FGFR3 signalling contribute to cetuximab resistance in wild-type KRAS tumour cells
- PMID: 22491422
- PMCID: PMC3326676
- DOI: 10.1038/bjc.2012.103
Cross-suppression of EGFR ligands amphiregulin and epiregulin and de-repression of FGFR3 signalling contribute to cetuximab resistance in wild-type KRAS tumour cells
Abstract
Background: In addition to the mutational status of KRAS, the epidermal growth factor receptor (EGFR) ligands amphiregulin (AREG) and epiregulin (EREG) might function as bona fide biomarkers of cetuximab (Ctx) sensitivity for most EGFR-driven carcinomas.
Methods: Lentivirus-delivered small hairpin RNAs were employed to specifically reduce AREG or EREG gene expression in wild-type KRAS A431 squamous cell carcinoma cells. Colony-forming assays were used to monitor the impact of AREG and EREG knockdown on Ctx efficacy. Amphiregulin and EREG protein expression levels were assessed by quantitative ELISA in parental A431 cells and in pooled populations of A431 cells adapted to grow in the presence of Ctx. A phosphoproteomic platform was used to measure the relative level of phosphorylation of 42 distinct receptor tyrosine kinases before and after the acquisition of resistance to Ctx.
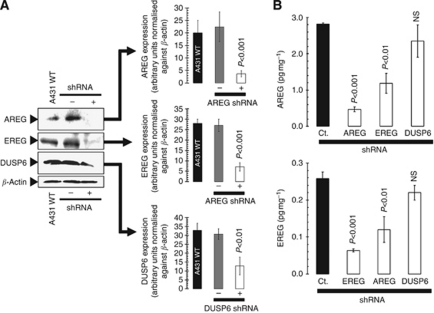
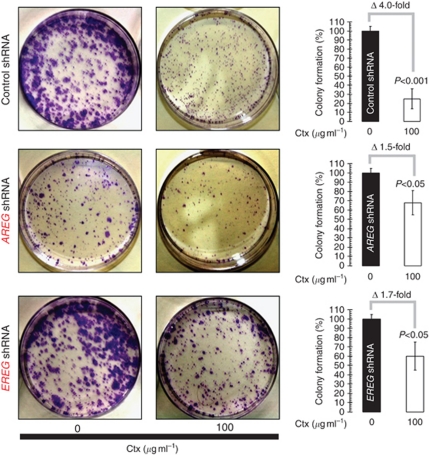
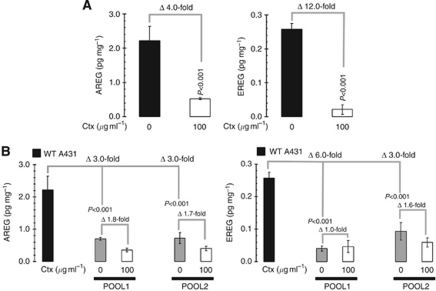
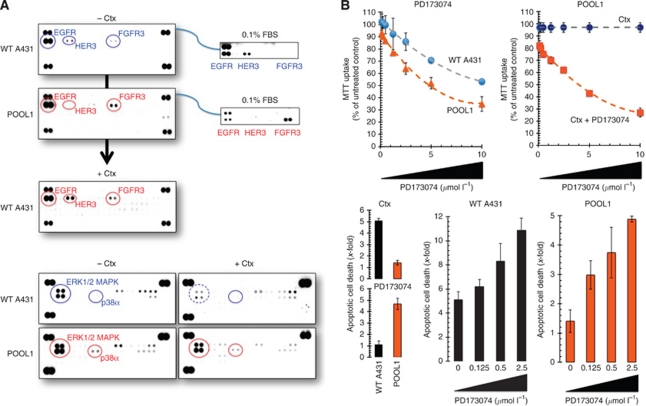
Results: Stable gene silencing of either ligand was found to notably reduce the expression of the other ligand. Parental A431 cells with normal expression levels of AREG/EREG exhibited significantly increased growth inhibition in response to Ctx, compared with derivatives that are engineered to produce minimal AREG/EREG. The parental A431 cells acutely treated with Ctx exhibited reduced basal expression levels of AREG/EREG. Pooled populations of Ctx-resistant A431 cells expressed significantly lower levels of AREG/EREG and were insensitive to the downregulatory effects of Ctx. Phosphoproteomic screen identified a remarkable hyperactivation of FGFR3 in Ctx-resistant A431 cells, which gained sensitivity to the cytotoxic and apoptotic effects of the FGFR3 TK inhibitor PD173074. The A431 parental cells acutely treated with Ctx rapidly activated FGFR3 and their concomitant exposure to Ctx and PD173074 resulted in synergistic apoptosis.
Conclusion: Cross-suppression of AREG/EREG expression may explain the tight co-expression of AREG and EREG, as well as their tendency to be more highly expressed than other EGFR ligands to determine Ctx efficacy. The positive selection for Ctx-resistant tumour cells exhibiting AREG/EREG cross-suppression may have an important role in the emergence of Ctx resistance. As de-repression of FGFR3 activity rapidly replaces the loss of EGFR-ligand signalling in terms of cell proliferation and survival, combinations of Ctx and FGFR3-targeted drugs may be a valuable strategy to enhance the efficacy of single Ctx while preventing or delaying acquired resistance to Ctx.
Conflict of interest statement
Cristina Oliveras-Ferraros received a research salary from a Grant Award by the ‘Fundacion Salud 2000’, which is promoted by Merck Serono (Madrid, Spain). All other authors declare no conflict of interest.
Figures
References
-
- Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP, Maiese K, Scher HI, Sliwkowski MX (2002) Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2: 127–137 - PubMed
-
- Baker JB, Dutta D, Watson D, Maddala T, Shak S, Rowinsky EK, Xu L, Clark E, Mauro DJ, Khambata-Ford S (2008) Evaluation of tumor gene expression and K-Ras mutations in FFPE tumor tissue as predictors of response to cetuximab in metastatic colorectal cancer. J Clin Oncol 27Suppl3512 (abstract)
-
- De Roock W, Jansenss M, Biesmans B, Jacobs J, De Schutter J, Fieuws E, Van Cutsem E, Tejpar S (2009) DUSPs as marker of MEK/Erk activation in primary colorectal cancer. J Clin Oncol 27: 15s (abstract 4064)
-
- Engelman JA, Janne PA (2008) Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 14: 2895–2899 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
