

G-protein-coupled receptor 30 mediates rapid neuroprotective effects of estrogen via depression of NR2B-containing NMDA receptors
- PMID: 22492045
- PMCID: PMC6620914
- DOI: 10.1523/JNEUROSCI.5828-11.2012
G-protein-coupled receptor 30 mediates rapid neuroprotective effects of estrogen via depression of NR2B-containing NMDA receptors
Abstract
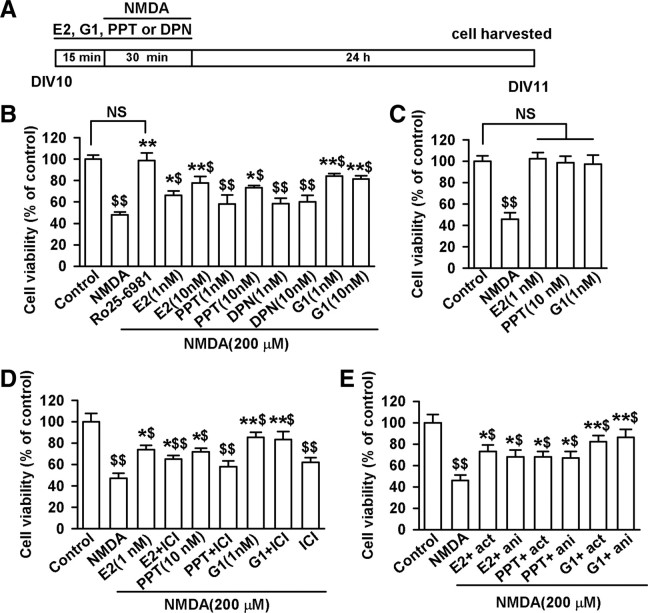
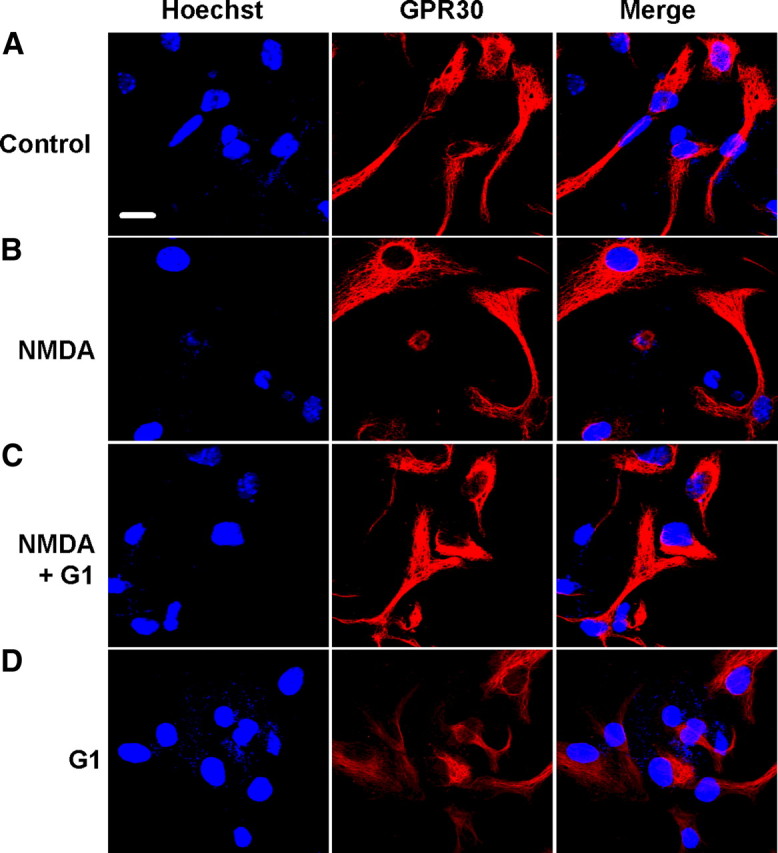
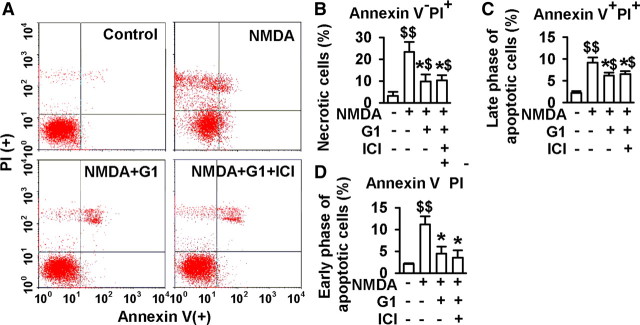
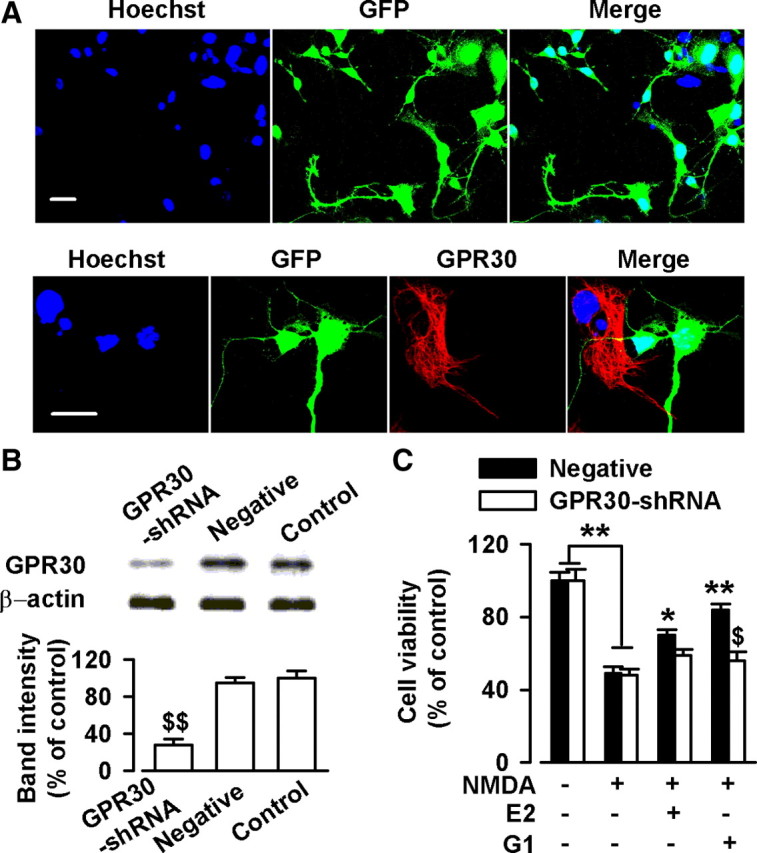
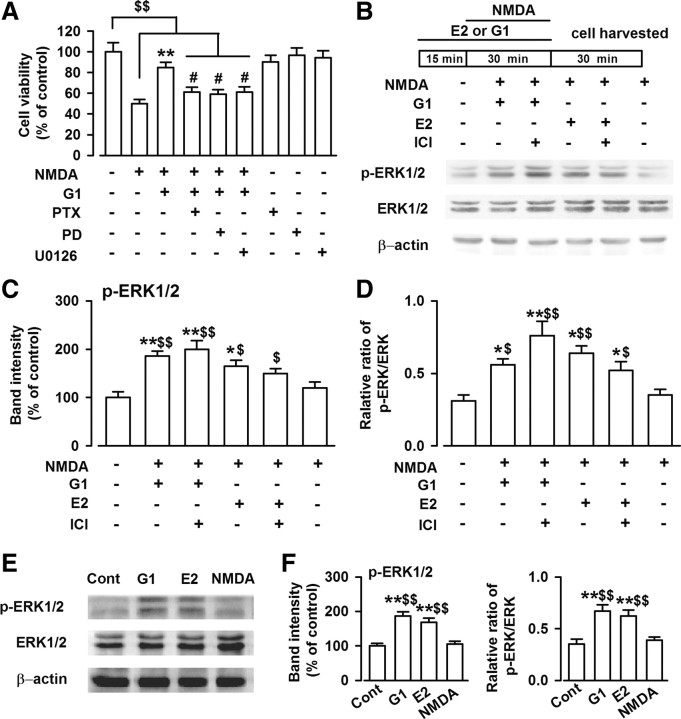
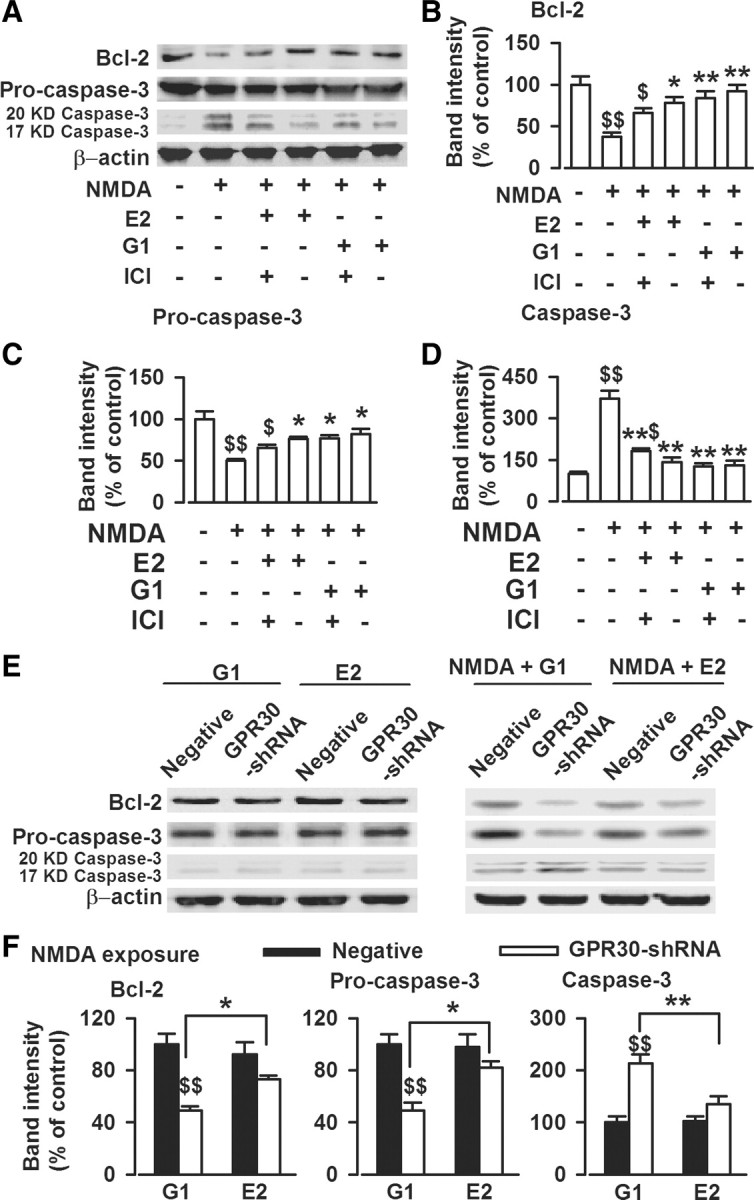
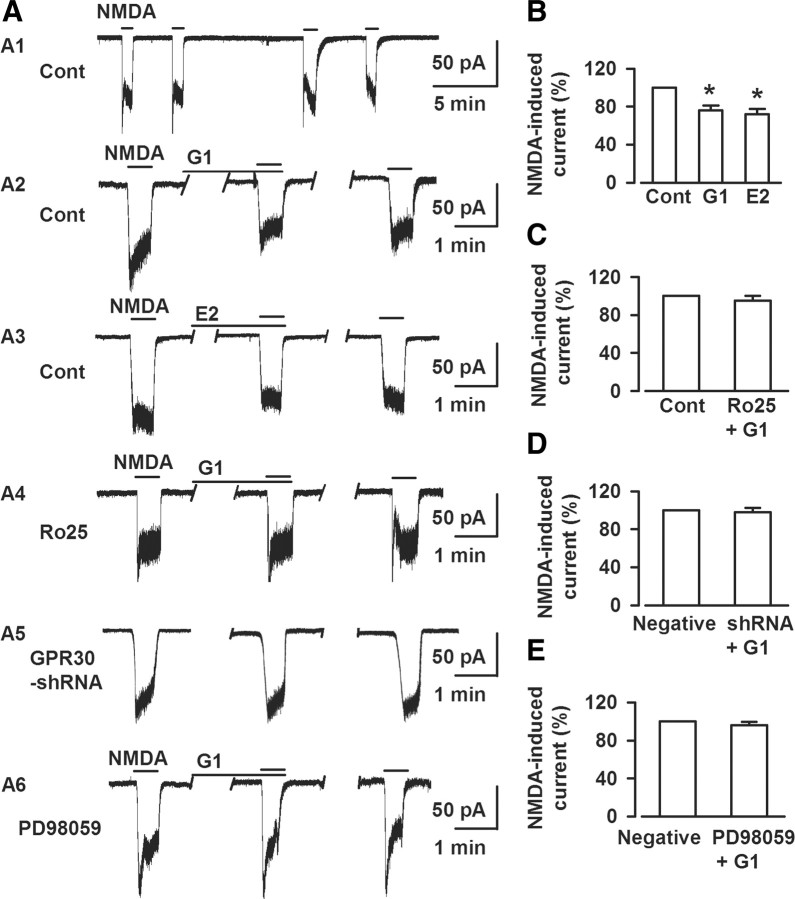
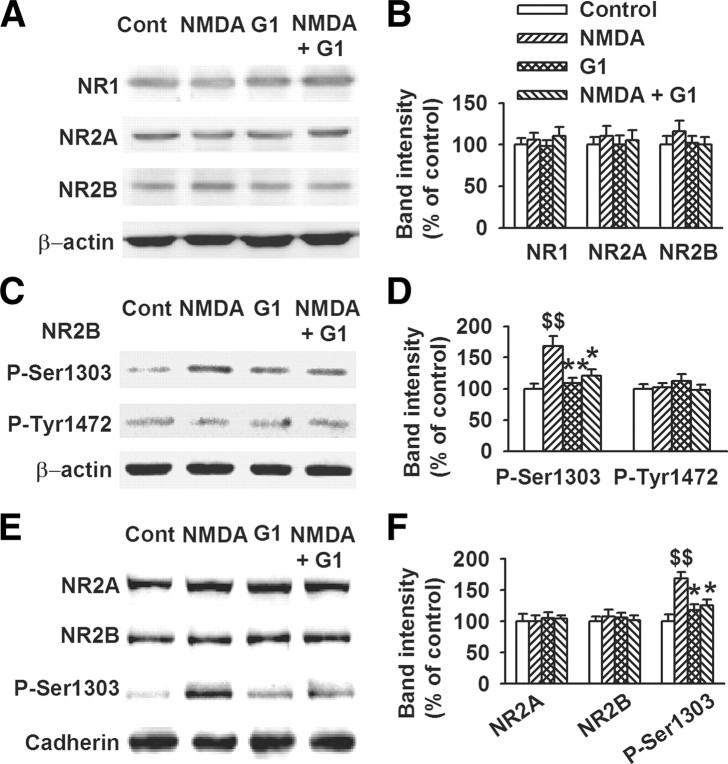
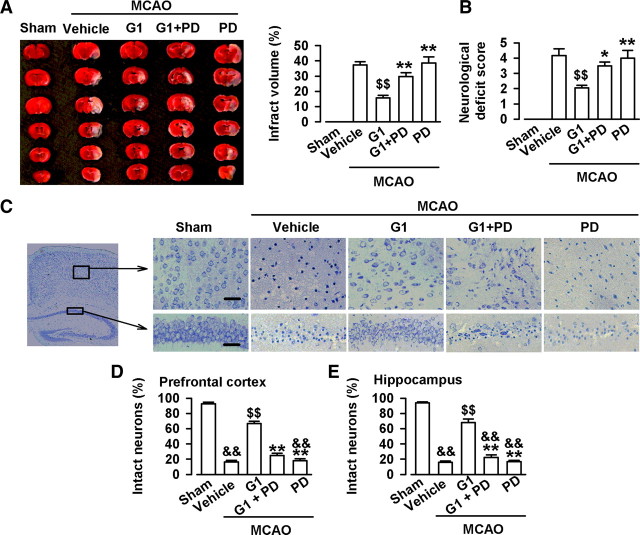
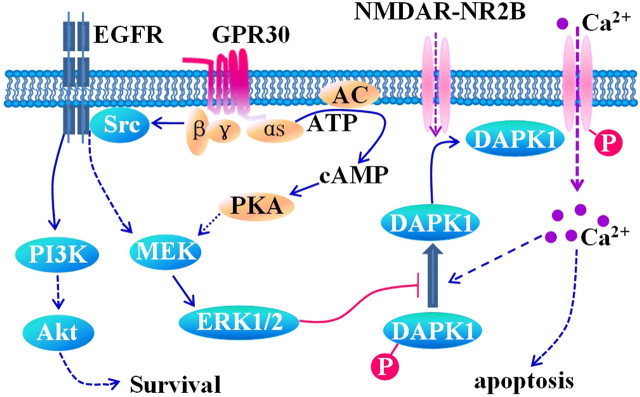
17-β-estradiol (E2) is a steroid hormone involved in neuroprotection against excitotoxicity and other forms of brain injury. Through genomic and nongenomic mechanisms, E2 modulates neuronal excitability and signal transmission by regulating NMDA and non-NMDA receptors. However, the mechanisms and identity of the receptors involved remain unclear, even though studies have suggested that estrogen G-protein-coupled receptor 30 (GPR30) is linked to protection against ischemic injury. In the culture cortical neurons, treatment with E2 and the GPR30 agonist G1 for 45 min attenuated the excitotoxicity induced by NMDA exposure. The acute neuroprotection mediated by GPR30 is dependent on G-protein-coupled signals and ERK1/2 activation, but independent on transcription or translation. Knockdown of GPR30 using short hairpin RNAs (shRNAs) significantly reduced the E2-induced rapid neuroprotection. Patch-clamp recordings revealed that GPR30 activation depressed exogenous NMDA-elicited currents. Short-term GPR30 activation did not affect the expression of either NR2A- or NR2B-containing NMDARs; however, it depressed NR2B subunit phosphorylation at Ser-1303 by inhibiting the dephosphorylation of death-associated protein kinase 1 (DAPK1). DAPK1 knockdown using shRNAs significantly blocked NR2B subunit phosphorylation at Ser-1303 and abolished the GPR30-mediated depression of exogenous NMDA-elicited currents. Lateral ventricle injection of the GPR30 agonist G1 (0.2 μg) provided significant neuroprotection in the ovariectomized female mice subjected to middle cerebral artery occlusion. These findings provide direct evidence that fast neuroprotection by estradiol is partially mediated by GPR30 and the subsequent downregulation of NR2B-containing NMDARs. The modulation of DAPK1 activity by GPR30 may be an important mediator of estradiol-dependent neuroprotection.
Figures

References
-
- Anjum R, Roux PP, Ballif BA, Gygi SP, Blenis J. The tumor suppressor DAP kinase is a target of RSK-mediated survival signaling. Curr Biol. 2005;15:1762–1767. - PubMed
-
- Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium. 2003;34:325–337. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous