

Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens
- PMID: 22492626
- PMCID: PMC3413110
- DOI: 10.1093/nar/gks299
Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens
Abstract
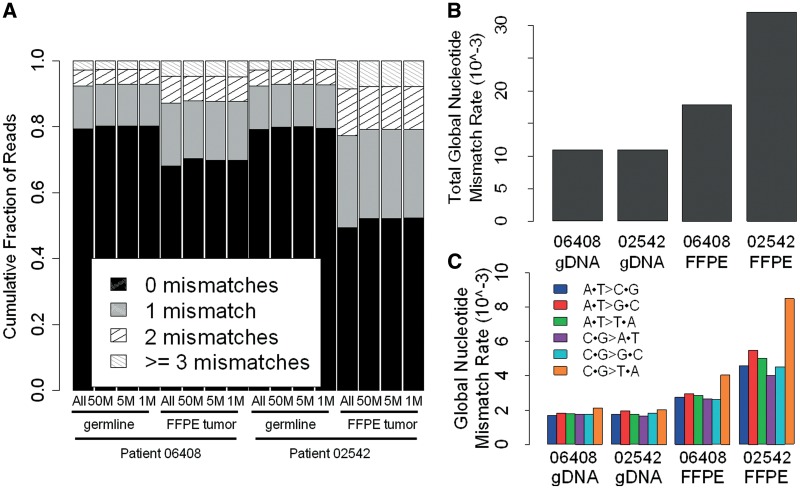
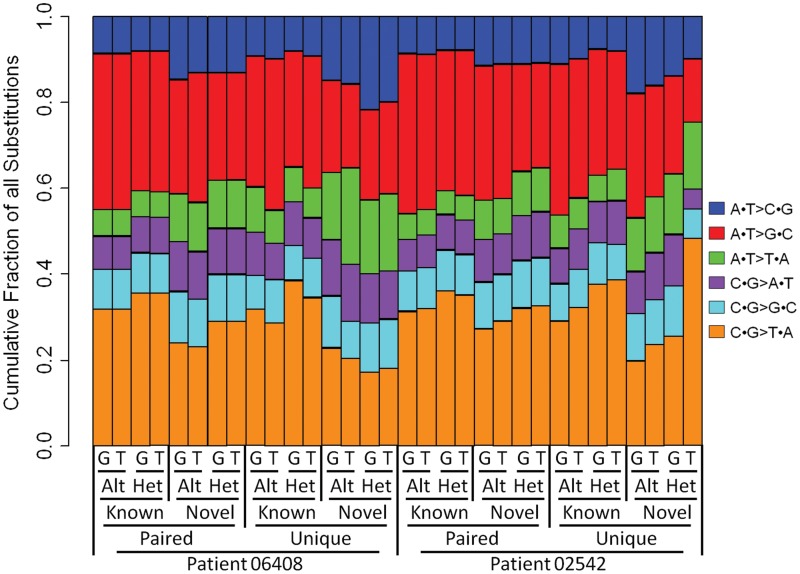
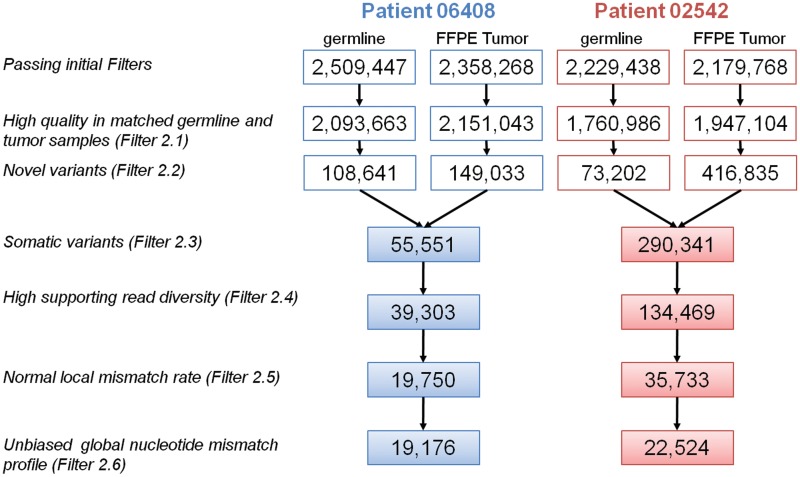
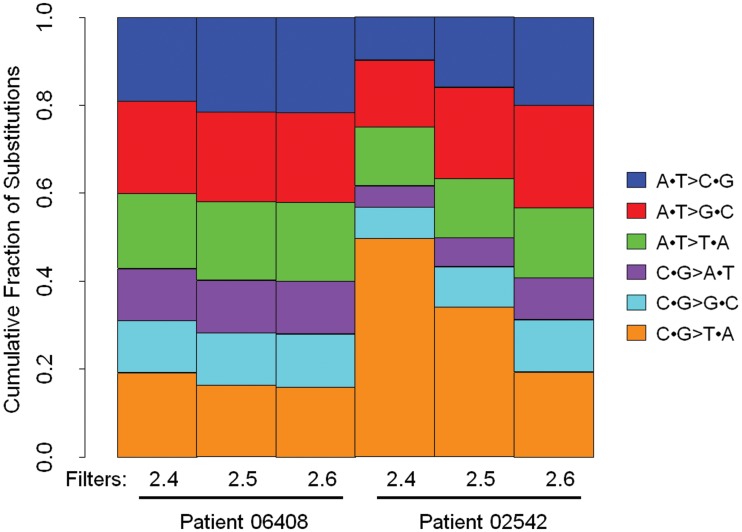
The utilization of archived, formalin-fixed paraffin-embedded (FFPE) tumor samples for massive parallel sequencing has been challenging due to DNA damage and contamination with normal stroma. Here, we perform whole genome sequencing of DNA isolated from two triple-negative breast cancer tumors archived for >11 years as 5 µm FFPE sections and matched germline DNA. The tumor samples show differing amounts of FFPE damaged DNA sequencing reads revealed as relatively high alignment mismatch rates enriched for C · G > T · A substitutions compared to germline samples. This increase in mismatch rate is observable with as few as one million reads, allowing for an upfront evaluation of the sample integrity before whole genome sequencing. By applying innovative quality filters incorporating global nucleotide mismatch rates and local mismatch rates, we present a method to identify high-confidence somatic mutations even in the presence of FFPE induced DNA damage. This results in a breast cancer mutational profile consistent with previous studies and revealing potentially important functional mutations. Our study demonstrates the feasibility of performing genome-wide deep sequencing analysis of FFPE archived tumors of limited sample size such as residual cancer after treatment or metastatic biopsies.
Figures
References
-
- Lee W, Jiang Z, Liu J, Haverty PM, Guan Y, Stinson J, Yue P, Zhang Y, Pant KP, Bhatt D, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2010;465:473–477. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
