

Inferring phylogenies from RAD sequence data
- PMID: 22493668
- PMCID: PMC3320897
- DOI: 10.1371/journal.pone.0033394
Inferring phylogenies from RAD sequence data
Abstract
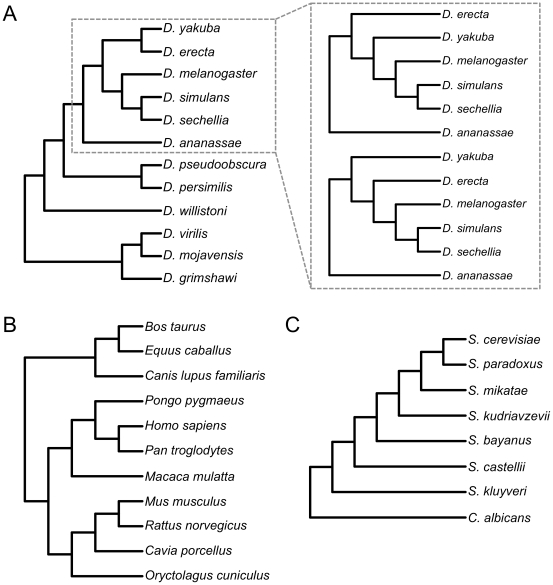
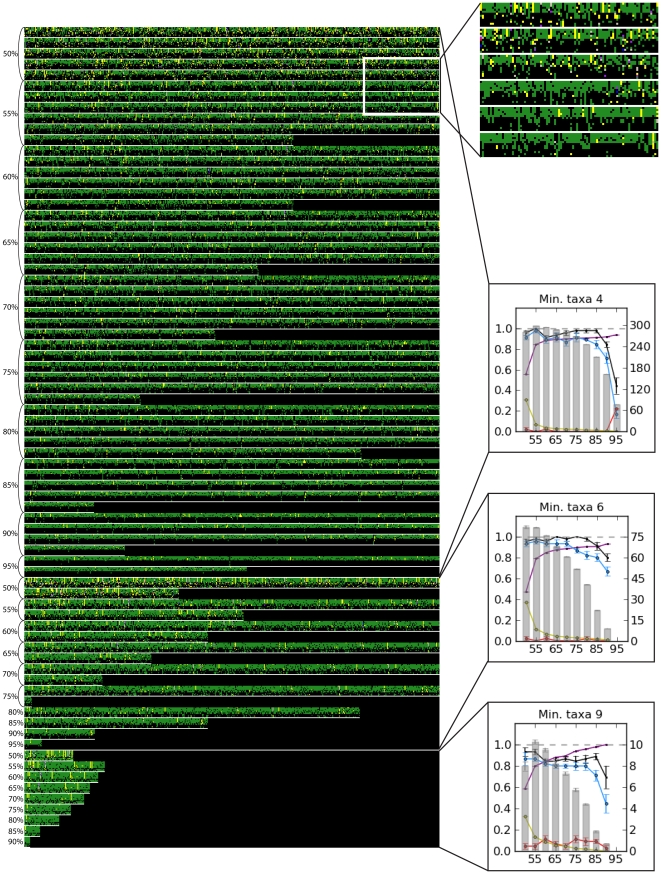
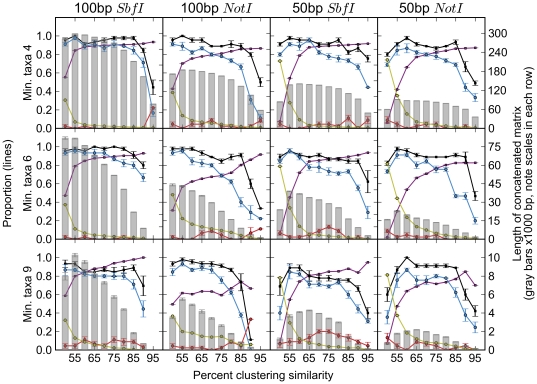
Reduced-representation genome sequencing represents a new source of data for systematics, and its potential utility in interspecific phylogeny reconstruction has not yet been explored. One approach that seems especially promising is the use of inexpensive short-read technologies (e.g., Illumina, SOLiD) to sequence restriction-site associated DNA (RAD)--the regions of the genome that flank the recognition sites of restriction enzymes. In this study, we simulated the collection of RAD sequences from sequenced genomes of different taxa (Drosophila, mammals, and yeasts) and developed a proof-of-concept workflow to test whether informative data could be extracted and used to accurately reconstruct "known" phylogenies of species within each group. The workflow consists of three basic steps: first, sequences are clustered by similarity to estimate orthology; second, clusters are filtered by taxonomic coverage; and third, they are aligned and concatenated for "total evidence" phylogenetic analysis. We evaluated the performance of clustering and filtering parameters by comparing the resulting topologies with well-supported reference trees and we were able to identify conditions under which the reference tree was inferred with high support. For Drosophila, whole genome alignments allowed us to directly evaluate which parameters most consistently recovered orthologous sequences. For the parameter ranges explored, we recovered the best results at the low ends of sequence similarity and taxonomic representation of loci; these generated the largest supermatrices with the highest proportion of missing data. Applications of the method to mammals and yeasts were less successful, which we suggest may be due partly to their much deeper evolutionary divergence times compared to Drosophila (crown ages of approximately 100 and 300 versus 60 Mya, respectively). RAD sequences thus appear to hold promise for reconstructing phylogenetic relationships in younger clades in which sufficient numbers of orthologous restriction sites are retained across species.
Conflict of interest statement
Figures
References
-
- Wu F, Mueller LA, Crouzillat D, Pétiard V, Tanksley SD. Combining bioinformatics and phylogenetics to identify large sets of single-copy orthologous genes (COSII) for comparative, evolutionary and systematic studies: A test case in the Euasterid plant clade. Genetics. 2006;174:1407–1420. - PMC - PubMed
-
- Rokas A, Williams BL, King N, Carrol SB. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425:798–804. - PubMed
-
- Drosophila 12 Genomes Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
