

Phylogenetic incongruence in E. coli O104: understanding the evolutionary relationships of emerging pathogens in the face of homologous recombination
- PMID: 22493677
- PMCID: PMC3320906
- DOI: 10.1371/journal.pone.0033971
Phylogenetic incongruence in E. coli O104: understanding the evolutionary relationships of emerging pathogens in the face of homologous recombination
Abstract
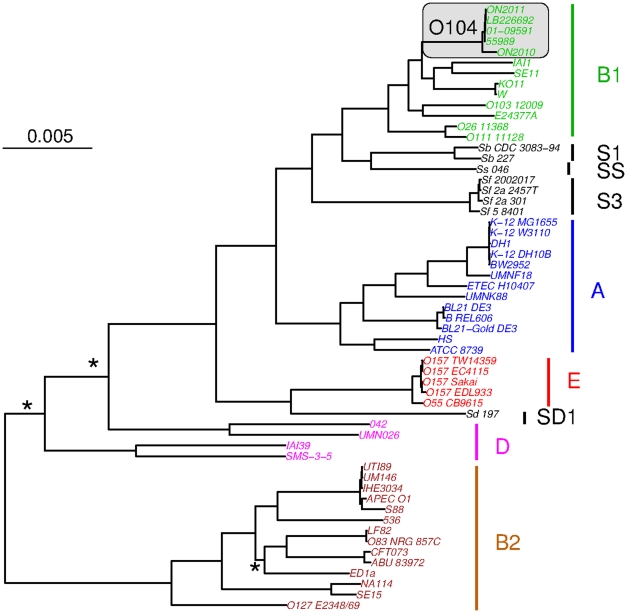
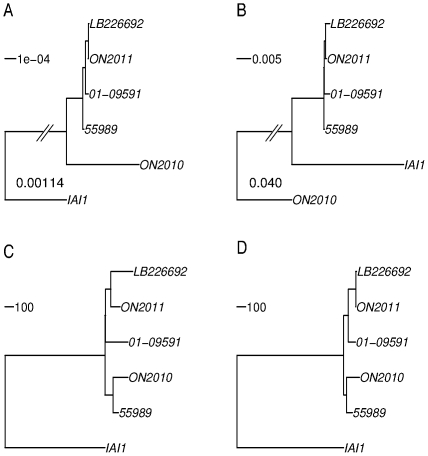
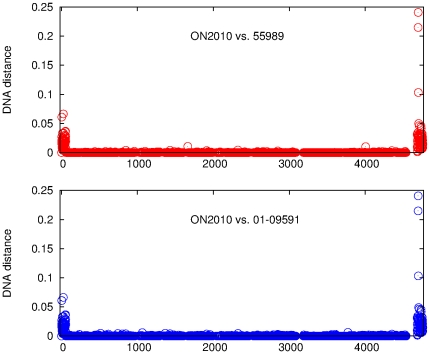
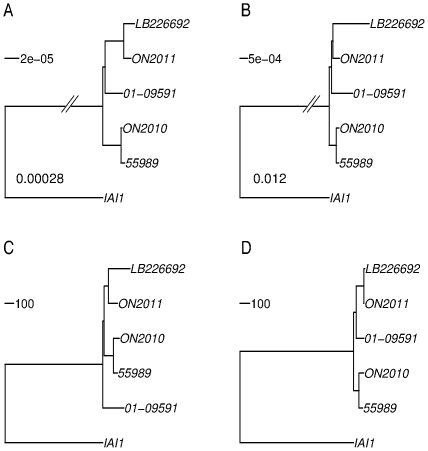
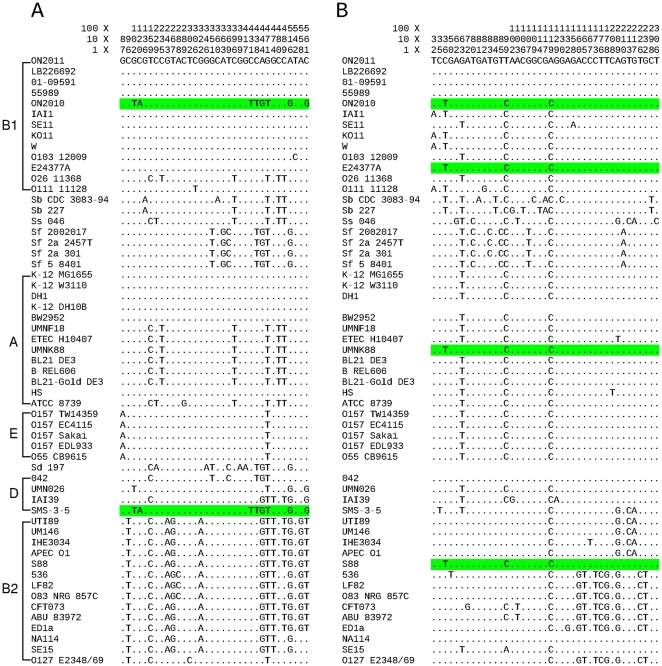
Escherichia coli O104:H4 was identified as an emerging pathogen during the spring and summer of 2011 and was responsible for a widespread outbreak that resulted in the deaths of 50 people and sickened over 4075. Traditional phenotypic and genotypic assays, such as serotyping, pulsed field gel electrophoresis (PFGE), and multilocus sequence typing (MLST), permit identification and classification of bacterial pathogens, but cannot accurately resolve relationships among genotypically similar but pathotypically different isolates. To understand the evolutionary origins of E. coli O104:H4, we sequenced two strains isolated in Ontario, Canada. One was epidemiologically linked to the 2011 outbreak, and the second, unrelated isolate, was obtained in 2010. MLST analysis indicated that both isolates are of the same sequence type (ST678), but whole-genome sequencing revealed differences in chromosomal and plasmid content. Through comprehensive phylogenetic analysis of five O104:H4 ST678 genomes, we identified 167 genes in three gene clusters that have undergone homologous recombination with distantly related E. coli strains. These recombination events have resulted in unexpectedly high sequence diversity within the same sequence type. Failure to recognize or adjust for homologous recombination can result in phylogenetic incongruence. Understanding the extent of homologous recombination among different strains of the same sequence type may explain the pathotypic differences between the ON2010 and ON2011 strains and help shed new light on the emergence of this new pathogen.
Conflict of interest statement
Figures






References
-
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. - PubMed
-
- Dobrindt U, Hacker J. Whole genome plasticity in pathogenic bacteria. Curr Opin Microbiol. 2001;4:550–557. - PubMed
-
- Hacker J, Blum-Oehler G, Muhldorfer I, Tschape H. Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol Microbiol. 1997;23:1089–1097. - PubMed
-
- McGraw EA, Li J, Selander RK, Whittam TS. Molecular evolution and mosaic structure of alpha, beta, and gamma intimins of pathogenic Escherichia coli. Mol Biol Evol. 1999;16:12–22. - PubMed
-
- Posada D, Crandall KA, Holmes EC. Recombination in evolutionary genomics. Annu Rev Genet. 2002;36:75–97. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
