

Structural disorder in eukaryotes
- PMID: 22496841
- PMCID: PMC3320622
- DOI: 10.1371/journal.pone.0034687
Structural disorder in eukaryotes
Abstract
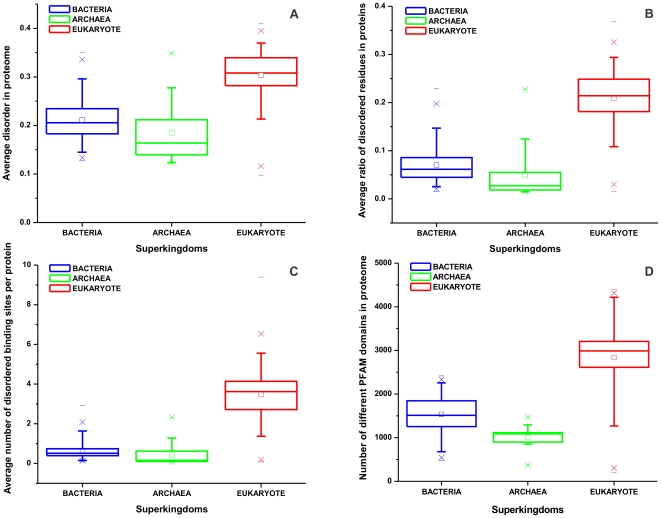
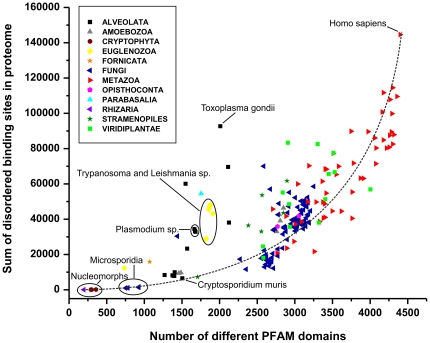
Based on early bioinformatic studies on a handful of species, the frequency of structural disorder of proteins is generally thought to be much higher in eukaryotes than in prokaryotes. To refine this view, we present here a comparative prediction study and analysis of 194 fully described eukaryotic proteomes and 87 reference prokaryotes for structural disorder. We found that structural disorder does distinguish eukaryotes from prokaryotes, but its frequency spans a very wide range in the two superkingdoms that largely overlap. The number of disordered binding regions and different Pfam domain types also contribute to distinguish eukaryotes from prokaryotes. Unexpectedly, the highest levels--and highest variability--of predicted disorder is found in protists, i.e. single-celled eukaryotes, often surpassing more complex eukaryote organisms, plants and animals. This trend contrasts with that of the number of domain types, which increases rather monotonously toward more complex organisms. The level of structural disorder appears to be strongly correlated with lifestyle, because some obligate intracellular parasites and endosymbionts have the lowest levels, whereas host-changing parasites have the highest level of predicted disorder. We conclude that protists have been the evolutionary hot-bed of experimentation with structural disorder, in a period when structural disorder was actively invented and the major functional classes of disordered proteins established.
Conflict of interest statement
Figures




References
-
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. - PubMed
-
- Tompa P. Unstructural biology coming of age. Curr Opin Struct Biol. 2011;21:419–425. - PubMed
-
- Tompa P, Szasz C, Buday L. Structural disorder throws new light on moonlighting. Trends Biochem Sci. 2005;30:484–489. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
