

Hypertelorism in Charcot-Marie-Tooth disease 1A from the common PMP22 duplication: A Case Report
- PMID: 22496945
- PMCID: PMC3321337
- DOI: 10.5001/omj.2012.34
Hypertelorism in Charcot-Marie-Tooth disease 1A from the common PMP22 duplication: A Case Report
Abstract
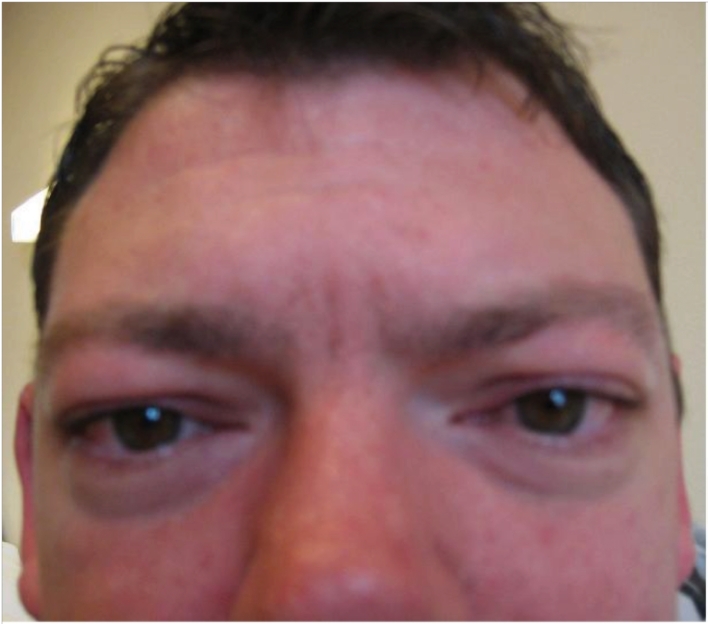
The 1.4Mb tandem-duplication in the PMP22 gene at 17p11.2 usually manifests as hereditary sensorimotor polyneuropathy with foot deformity, sensorineural hearing-loss, moderate developmental delay, and gait disturbance. Hypertelorism and marked phenotypic variability within a single family has not been reported. In a single family, the PMP22 tandem-duplication manifested as short stature, sensorimotor polyneuropathy, tremor, ataxia, sensorineural hearing-loss, and hypothyroidism in the 27 years-old index case, as mild facial dysmorphism, muscle cramps, tinnitus, intention tremor, bradydiadochokinesia, and sensorimotor polyneuropathy in the 31 year-old half-brother of the index-patient, and as sensorimotor polyneuropathy and foot-deformity in the father of the two. The half-brother additionally presented with hypertelorism, not previously reported in PMP22 tandem-duplication carriers. The presented cases show that the tandem-duplication 17p11.2 may present with marked intra-familial phenotype variability and that mild facial dysmorphism with stuck-out ears and hypertelorism may be a rare phenotypic feature of this mutation. The causal relation between facial dysmorphism and the PMP22 tandem-duplication, however, remains speculative.
Keywords: Hereditary neuropathy; Nerve conduction; Neuromuscular disorder; Peripheral nervous system.
Figures
References
-
- Moog U, Engelen JJ, Weber BW, Van Gelderen M, Steyaert J, Baas F, et al. Hereditary motor and sensory neuropathy (HMSN) IA, developmental delay and autism related disorder in a boy with duplication (17)(p11.2p12). Genet Couns 2004;15(1):73-80 - PubMed
-
- Bayani L, Sadeghi Tari A, Hamzeh-Doost K, Kasaii A. The bony interorbital distance and orbital measurements in the Iranian population: A CT study. Ir J Rad 2006;3:173-178
-
- Badano JL, Inoue K, Katsanis N, Lupski JR. New polymorphic short tandem repeats for PCR-based Charcot-Marie-Tooth disease type 1A duplication diagnosis. Clin Chem 2001. May;47(5):838-843 - PubMed
Publication types
LinkOut - more resources
Full Text Sources