

Long-term epilepsy-associated tumors
- PMID: 22497610
- PMCID: PMC8029234
- DOI: 10.1111/j.1750-3639.2012.00582.x
Long-term epilepsy-associated tumors
Abstract
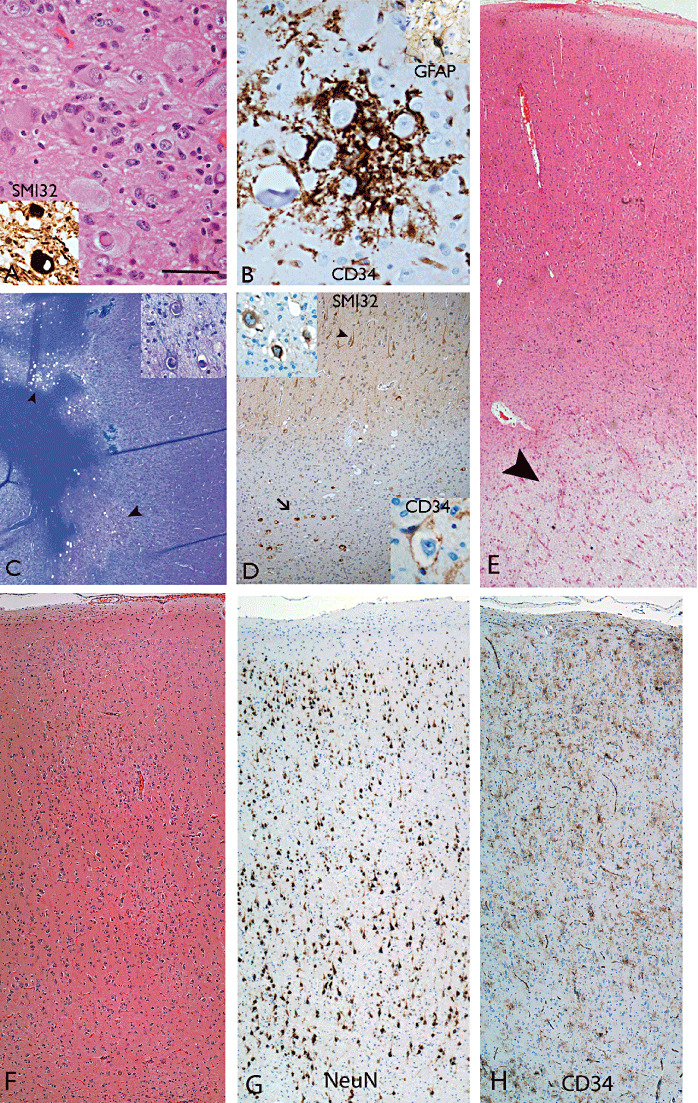
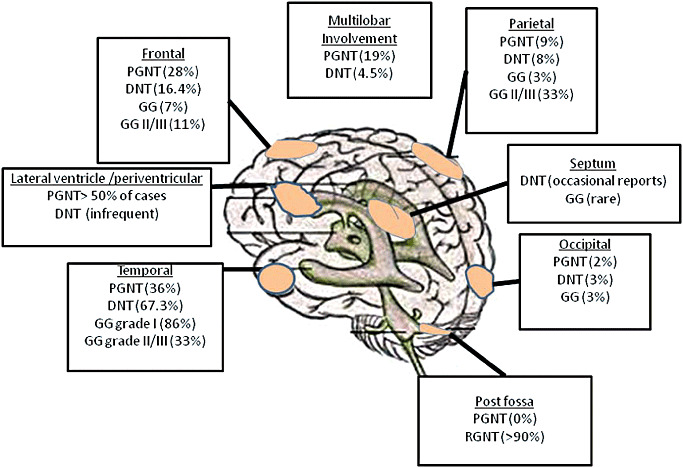
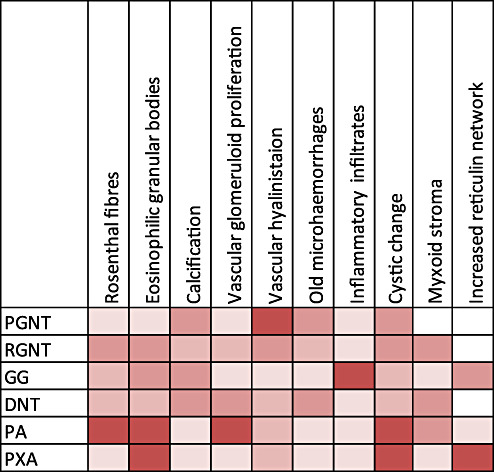
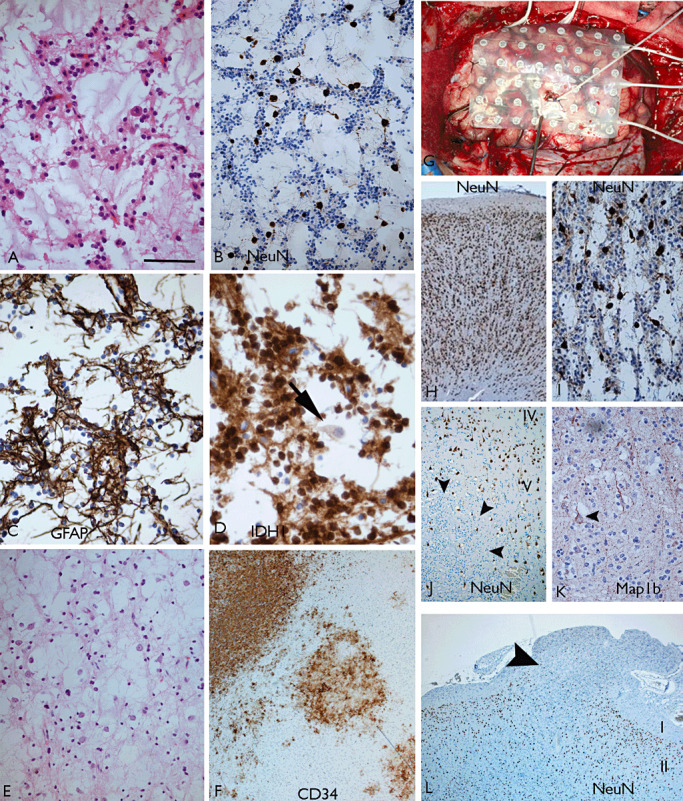
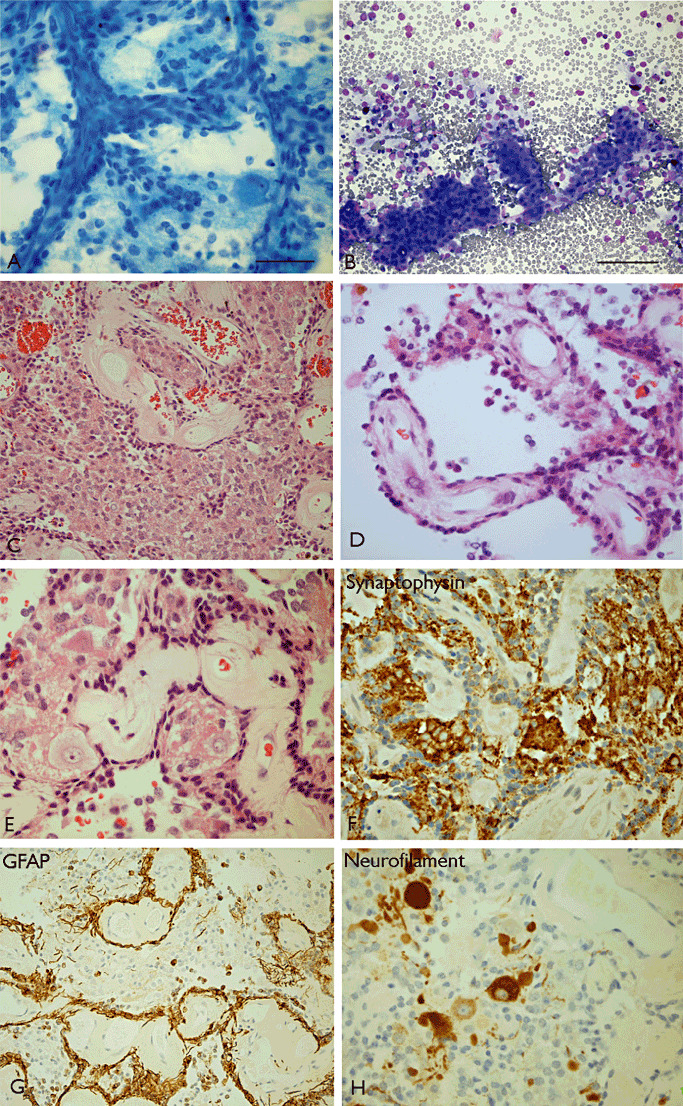
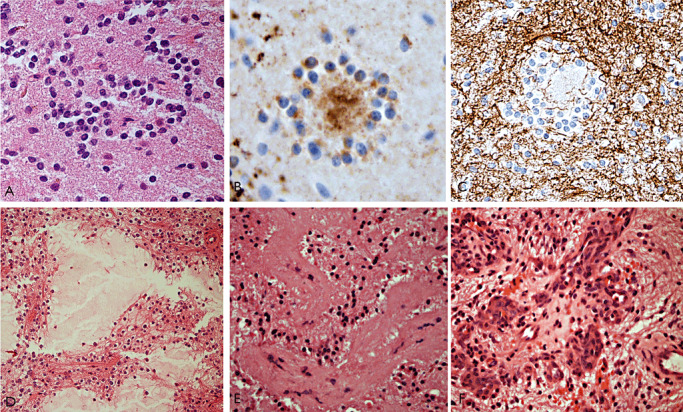
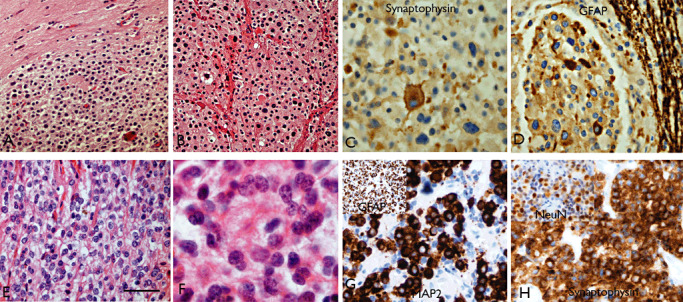
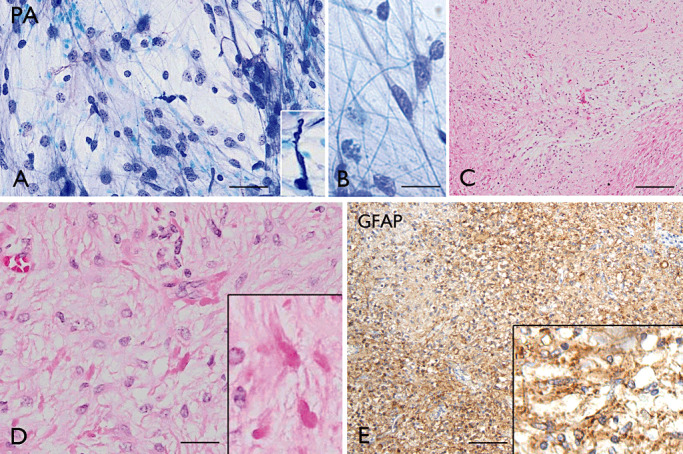
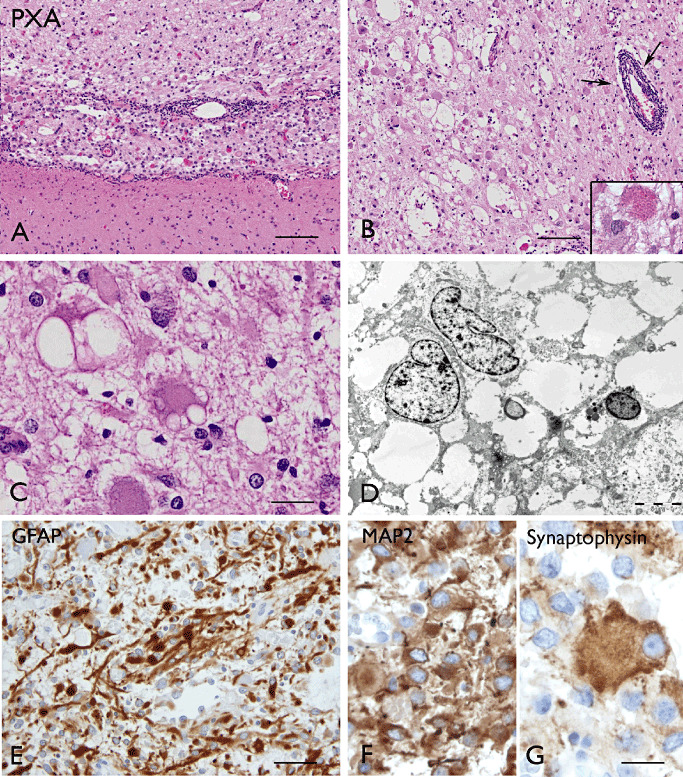
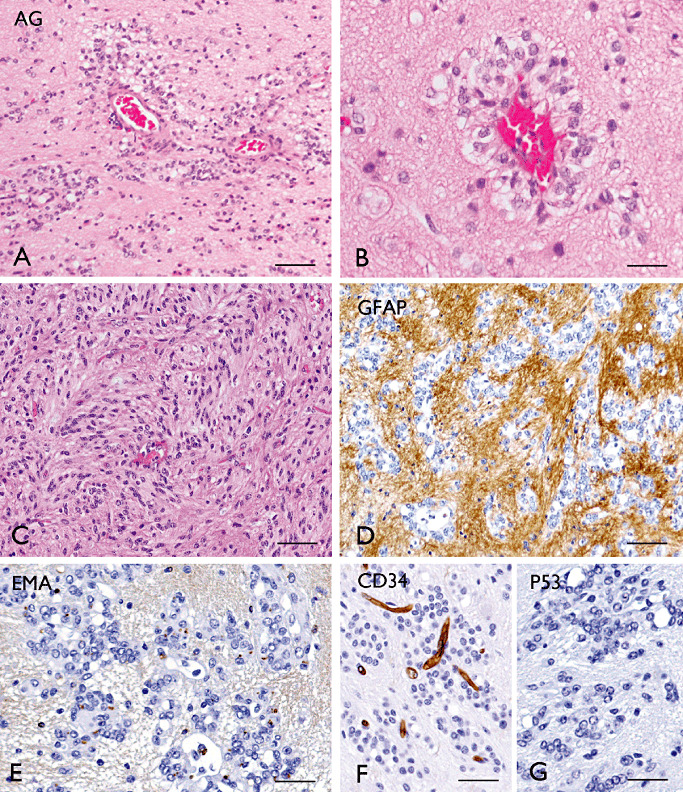
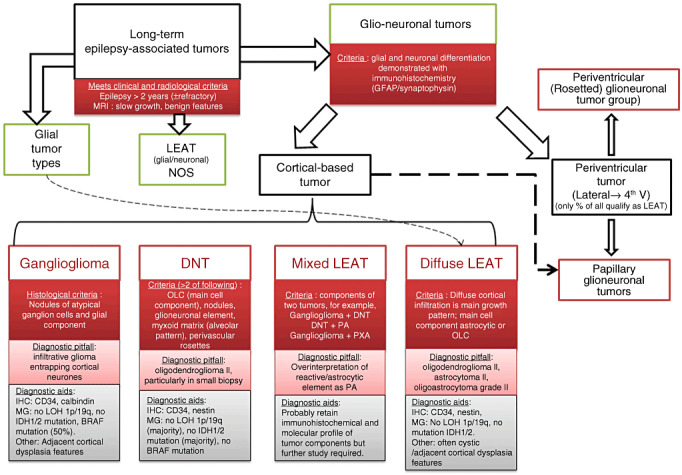
The term long-term epilepsy associated tumor (LEAT) encompasses lesions identified in patients investigated for long histories (often 2 years or more) of drug-resistant epilepsy. They are generally slowly growing, low grade, cortically based tumors, more often arising in younger age groups and in many cases exhibit neuronal in addition to glial differentiation. Gangliogliomas and dysembryoplastic neuroepithelial tumors predominate in this group. LEATs are further united by cyto-architectural changes that may be present in the adjacent cortex which have some similarities to developmental focal cortical dysplasias (FCD); these are now grouped as FCD type IIIb in the updated International League Against Epilepsy (ILAE) classification. In the majority of cases, surgical treatments are beneficial from both perspectives of managing the seizures and the tumor. However, in a minority, seizures may recur, tumors may show regrowth or recurrence, and rarely undergo anaplastic progression. Predicting and identifying tumors likely to behave less favorably are key objectives of the neuropathologist. With immunohistochemistry and modern molecular pathology, it is becoming increasingly possible to refine diagnostic groups. Despite this, some LEATs remain difficult to classify, particularly tumors with "non-specific" or diffuse growth patterns. Modification of LEAT classification is inevitable with the goal of unifying terminological criteria applied between centers for accurate clinico-pathological-molecular correlative data to emerge. Finally, establishing the epileptogenic components of LEAT, either within the lesion or perilesional cortex, will elucidate the cellular mechanisms of epileptogenesis, which in turn will guide optimal surgical management of these lesions.
© 2012 The Authors; Brain Pathology © 2012 International Society of Neuropathology.
Figures
References
-
- Adam C, Polivka M, Carpentier A, George B, Gray F (2007) Papillary glioneuronal tumor: not always a benign tumor? Clin Neuropathol 26:119–124. - PubMed
-
- Agarwal S, Suri V, Rishi A, Shukla B, Garg A, Sharma MC et al (2009) Glioneuronal tumor with neuropil‐like islands: a new entity. Neuropathology 29:96–100. - PubMed
-
- Aldape K, Burger PC, Perry A (2007) Clinicopathologic aspects of 1p/19q loss and the diagnosis of oligodendroglioma. Arch Pathol Lab Med 131:242–251. - PubMed
-
- Anan M, Inoue R, Ishii K, Abe T, Fujiki M, Kobayashi H et al (2009) A rosette‐forming glioneuronal tumor of the spinal cord: the first case of a rosette‐forming glioneuronal tumor originating from the spinal cord. Hum Pathol 40:898–901. - PubMed
-
- Aronica E, Crino PB (2011) Inflammation in epilepsy: clinical observations. Epilepsia 52(Suppl. 3):26–32. - PubMed
