

Comment
doi: 10.1016/j.cub.2012.02.056.
Axon degeneration: where the Wlds things are
Affiliations
- PMID: 22497934
- PMCID: PMC4449840
- DOI: 10.1016/j.cub.2012.02.056
Item in Clipboard
Comment
Axon degeneration: where the Wlds things are
Curr Biol.
.
Abstract
Expression of the Wld(s) protein significantly delays axon degeneration in injuries and diseases, but the mechanism for this protection is unknown. Two recent reports present evidence that axonal mitochondria are required for Wld(S)-mediated axon protection.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures
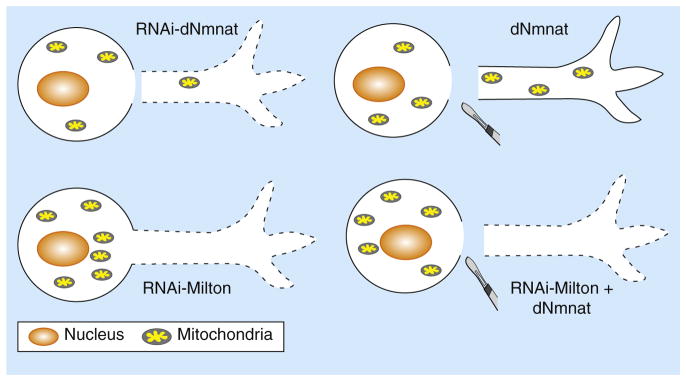
The Drosophila protein dNmnat is necessary and sufficient for
axonal survival, as RNAi-mediated knockdown of endogenous dNmnat induces
spontaneous axon degeneration and depletion of mitochondria in the axon (top
left), while upregulation of dNmnat delays axon degeneration from axotomy and
rescues the depletion of axonal mitochondria (top right). Blocking mitochondrial
entry into the axon by knocking down Milton, a linker protein required for
anterograde mitochondrial transport, leads to spontaneous axon degeneration
(bottom left). This degeneration cannot be rescued even with upregulation of
dNmnat (bottom right), indicating that axonal mitochondria are required for
dNmnat-mediated maintenance of normal axon survival and axon protection
following nerve injuries.
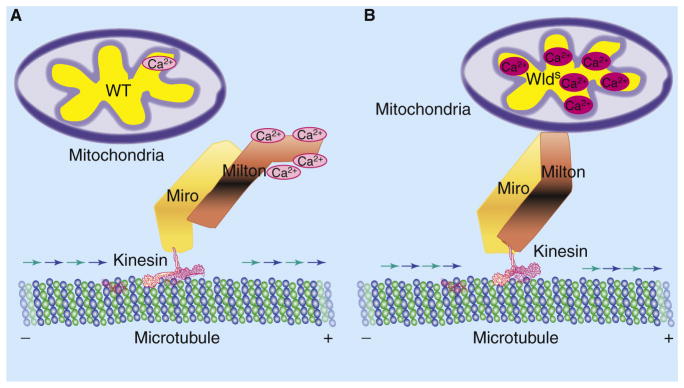
(A) In injured wild-type (WT) axons, an influx of extracellular
Ca2+ exceeds the threshold of mitochondrial
Ca2+ buffering and leads to a local, transient increase
of Ca2+ in the axon. Ca2+ binding to the
Milton–Miro protein complex leads to undocking of mitochondria from the
kinesin-dependent anterograde transport machinery, thereby decreasing
mitochondrial movement. (B) In injured WldS axons, increased
Ca2+ buffering by mitochondria decreases available free
Ca2+ to bind to the Milton–Miro complex, thus
maintaining the attachment of mitochondria to the anterograde transport
machinery and preserving motility of mitochondria in the axon.
Comment on
-
A novel Drosophila model of nerve injury reveals an essential role of Nmnat in maintaining axonal integrity.Curr Biol. 2012 Apr 10;22(7):590-5. doi: 10.1016/j.cub.2012.01.065. Epub 2012 Mar 15. Curr Biol. 2012. PMID: 22425156 Free PMC article.
-
WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering.Curr Biol. 2012 Apr 10;22(7):596-600. doi: 10.1016/j.cub.2012.02.043. Epub 2012 Mar 15. Curr Biol. 2012. PMID: 22425157 Free PMC article.
References
-
- Deckwerth TL, Johnson EM., Jr Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis) Dev Biol. 1994;165:63–72. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
