

Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice
- PMID: 22500629
- PMCID: PMC8822597
- DOI: 10.1016/j.neuron.2012.03.009
Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice
Abstract
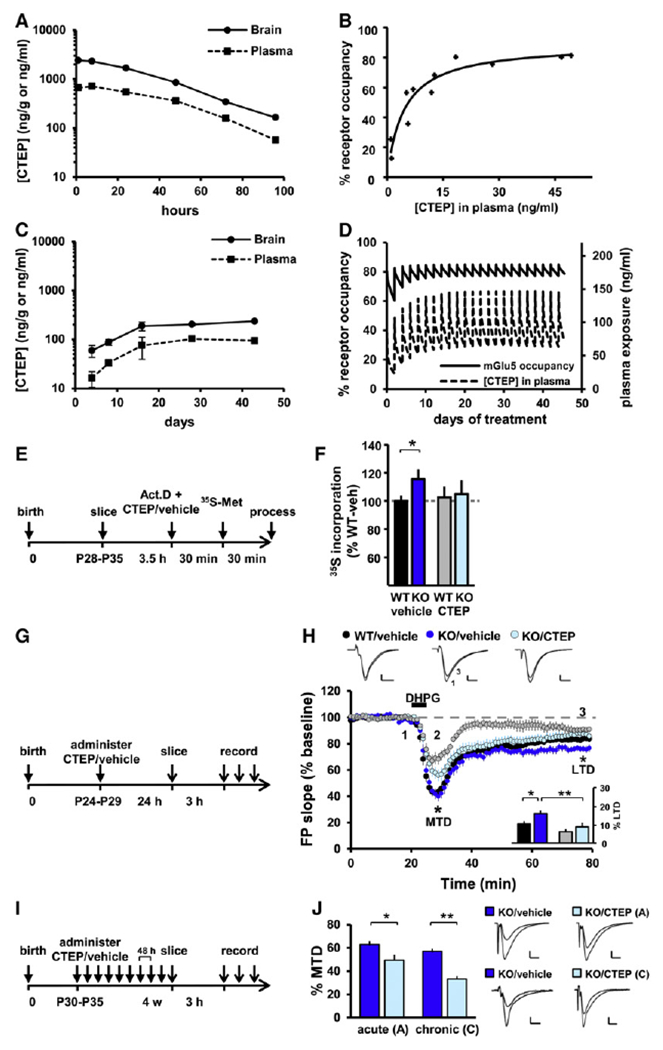
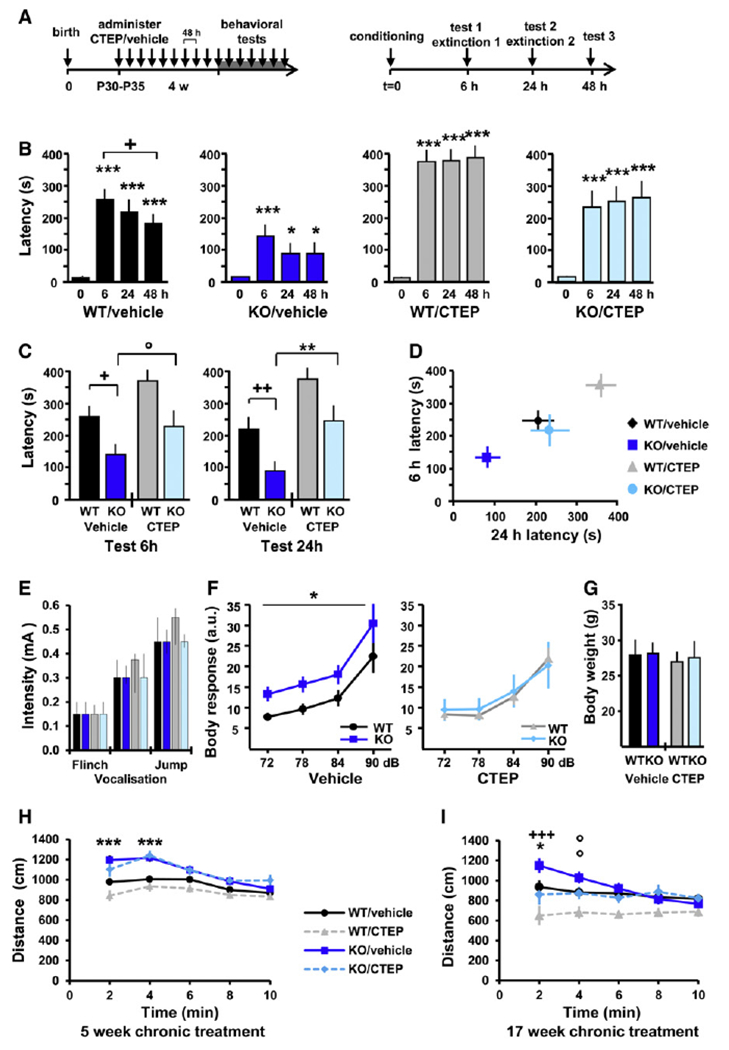
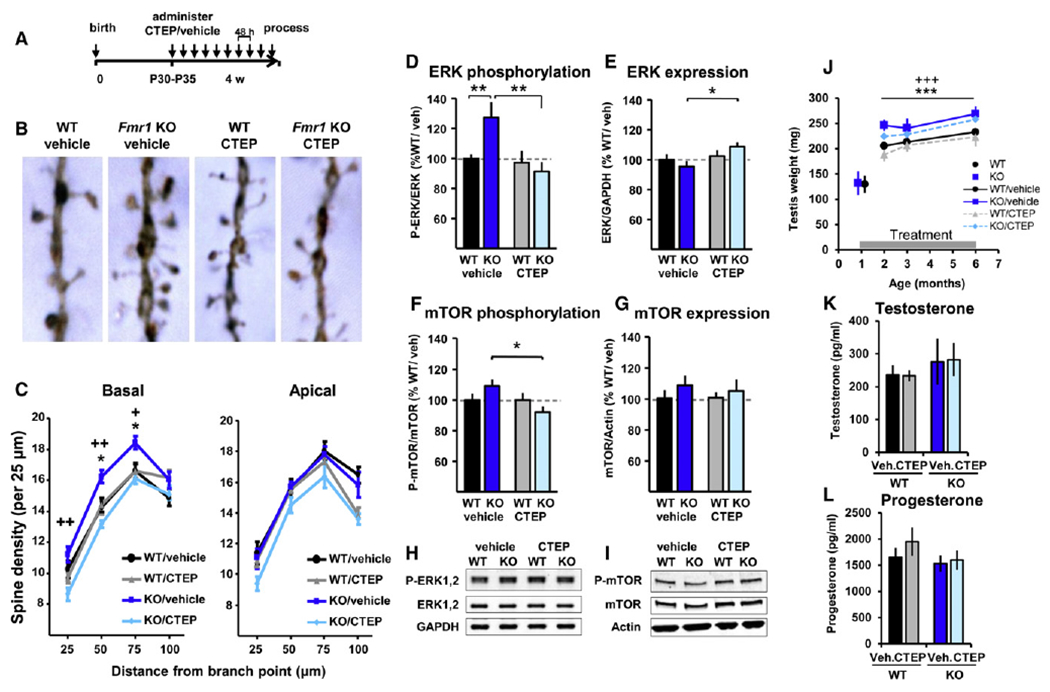
Fragile X syndrome (FXS) is the most common form of inherited intellectual disability. Previous studies have implicated mGlu5 in the pathogenesis of the disease, but a crucial unanswered question is whether pharmacological mGlu5 inhibition is able to reverse an already established FXS phenotype in mammals. Here we have used the novel, potent, and selective mGlu5 inhibitor CTEP to address this issue in the Fmr1 knockout mouse. Acute CTEP treatment corrects elevated hippocampal long-term depression, protein synthesis, and audiogenic seizures. Chronic treatment that inhibits mGlu5 within a receptor occupancy range of 81% ± 4% rescues cognitive deficits, auditory hypersensitivity, aberrant dendritic spine density, overactive ERK and mTOR signaling, and partially corrects macroorchidism. This study shows that a comprehensive phenotype correction in FXS is possible with pharmacological intervention starting in young adulthood, after development of the phenotype. It is of great interest how these findings may translate into ongoing clinical research testing mGlu5 inhibitors in FXS patients.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Fragile X syndrome therapeutics S(C)TEP through the developmental window.Neuron. 2012 Apr 12;74(1):1-3. doi: 10.1016/j.neuron.2012.03.014. Neuron. 2012. PMID: 22500622
-
Neurodevelopmental disorders: Reversing the fragile X phenotype.Nat Rev Neurosci. 2012 May 3;13(6):360. doi: 10.1038/nrn3255. Nat Rev Neurosci. 2012. PMID: 22551664 No abstract available.
-
Neurodevelopmental disorders: Glutamate blockers show benefit in models of autism spectrum disorders.Nat Rev Drug Discov. 2012 Jun 1;11(6):440-1. doi: 10.1038/nrd3761. Nat Rev Drug Discov. 2012. PMID: 22653211 No abstract available.
References
-
- Bear MF, Huber KM, and Warren ST (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. - PubMed
-
- Dalby KN, Morrice N, Caudwell FB, Avruch J, and Cohen P (1998). Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J. Biol. Chem 273, 1496–1505. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
