

The evolution of Dscam genes across the arthropods
- PMID: 22500922
- PMCID: PMC3364881
- DOI: 10.1186/1471-2148-12-53
The evolution of Dscam genes across the arthropods
Abstract
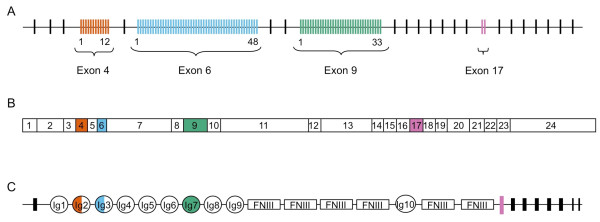
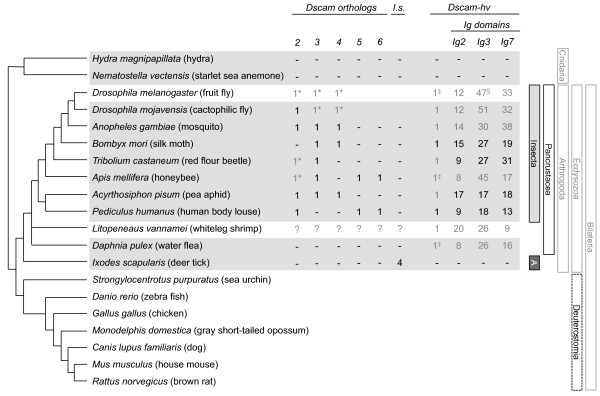
Background: One way of creating phenotypic diversity is through alternative splicing of precursor mRNAs. A gene that has evolved a hypervariable form is Down syndrome cell adhesion molecule (Dscam-hv), which in Drosophila melanogaster can produce thousands of isoforms via mutually exclusive alternative splicing. The extracellular region of this protein is encoded by three variable exon clusters, each containing multiple exon variants. The protein is vital for neuronal wiring where the extreme variability at the somatic level is required for axonal guidance, and it plays a role in immunity where the variability has been hypothesised to relate to recognition of different antigens. Dscam-hv has been found across the Pancrustacea. Additionally, three paralogous non-hypervariable Dscam-like genes have also been described for D. melanogaster. Here we took a bioinformatics approach, building profile Hidden Markov Models to search across species for putative orthologs to the Dscam genes and for hypervariable alternatively spliced exons, and inferring the phylogenetic relationships among them. Our aims were to examine whether Dscam orthologs exist outside the Bilateria, whether the origin of Dscam-hv could lie outside the Pancrustacea, when the Dscam-like orthologs arose, how many alternatively spliced exons of each exon cluster were present in the most common recent ancestor, and how these clusters evolved.
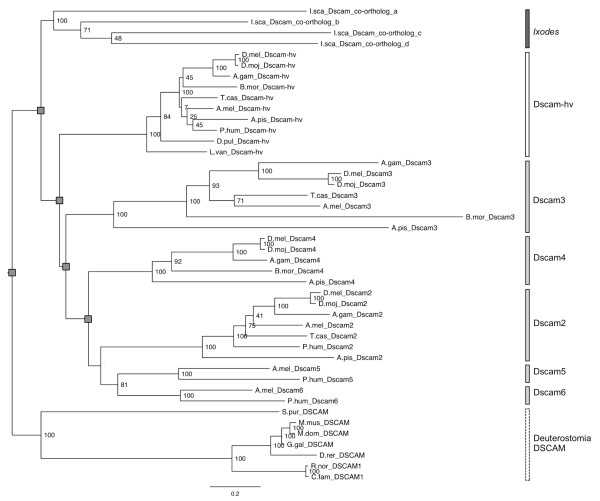
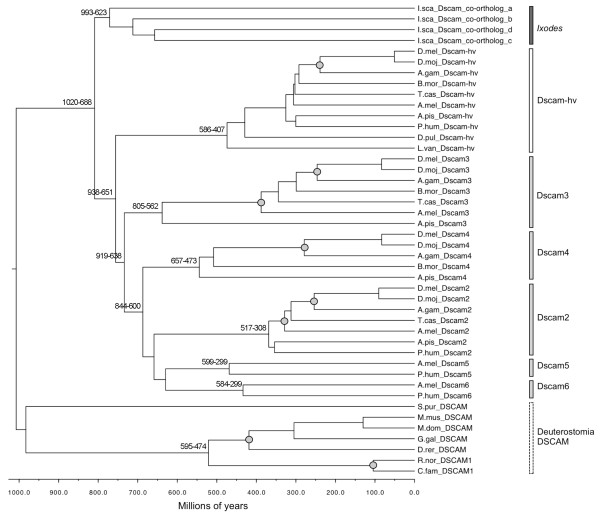
Results: Our results suggest that the origin of Dscam genes may lie after the split between the Cnidaria and the Bilateria and supports the hypothesis that Dscam-hv originated in the common ancestor of the Pancrustacea. Our phylogeny of Dscam gene family members shows six well-supported clades: five containing Dscam-like genes and one containing all the Dscam-hv genes, a seventh clade contains arachnid putative Dscam genes. Furthermore, the exon clusters appear to have experienced different evolutionary histories.
Conclusions: Dscam genes have undergone independent duplication events in the insects and in an arachnid genome, which adds to the more well-known tandem duplications that have taken place within Dscam-hv genes. Therefore, two forms of gene expansion seem to be active within this gene family. The evolutionary history of this dynamic gene family will be further unfolded as genomes of species from more disparate groups become available.
Figures
References
-
- C. elegans Sequencing Consortium. Genome Sequence of the Nematode C. elegans: A Platform for Investigating Biology. Science. 1998;282:2012–2018. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
