

Pangenomic study of Corynebacterium diphtheriae that provides insights into the genomic diversity of pathogenic isolates from cases of classical diphtheria, endocarditis, and pneumonia
- PMID: 22505676
- PMCID: PMC3370855
- DOI: 10.1128/JB.00183-12
Pangenomic study of Corynebacterium diphtheriae that provides insights into the genomic diversity of pathogenic isolates from cases of classical diphtheria, endocarditis, and pneumonia
Abstract
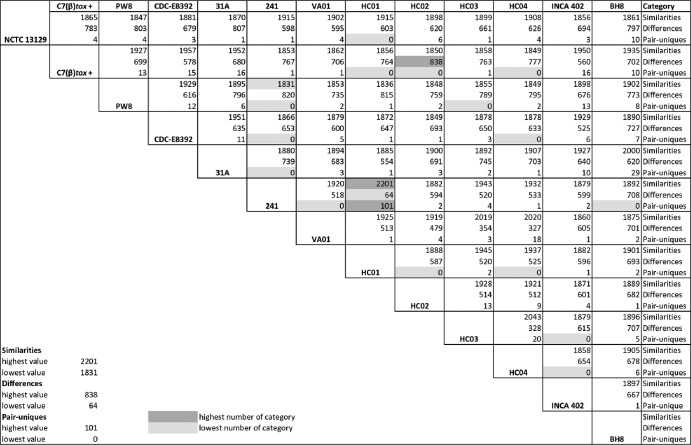
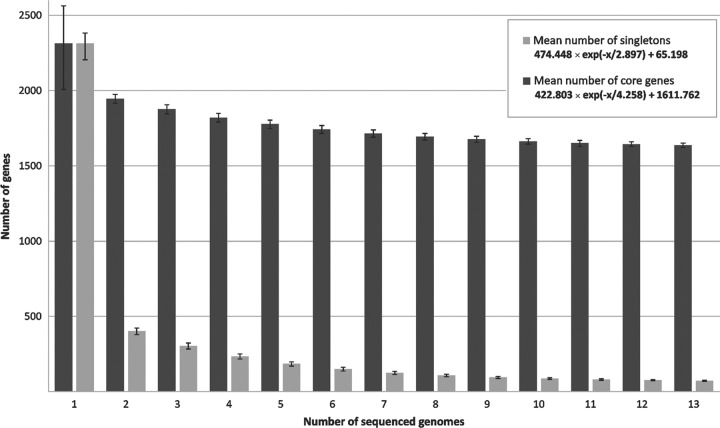
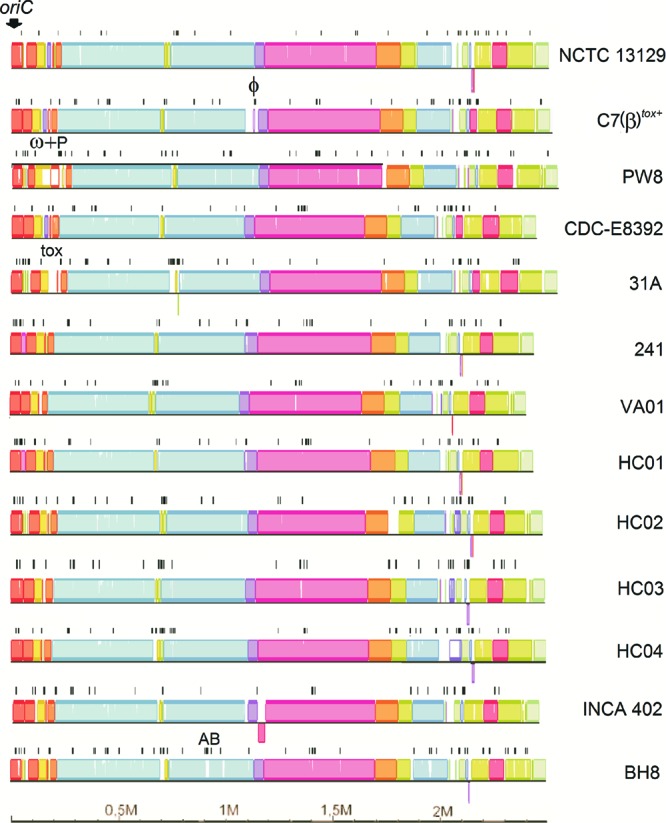
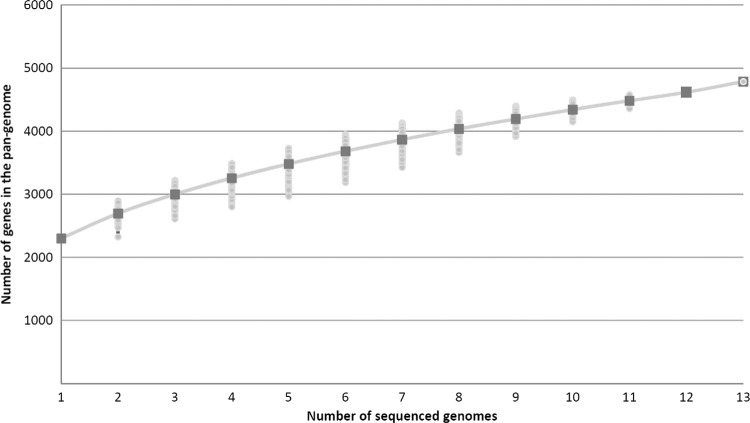
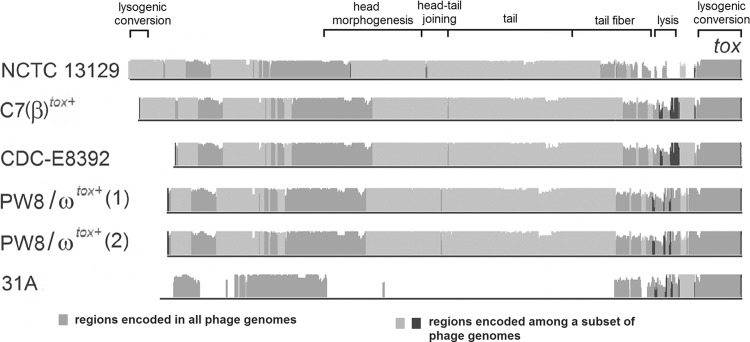
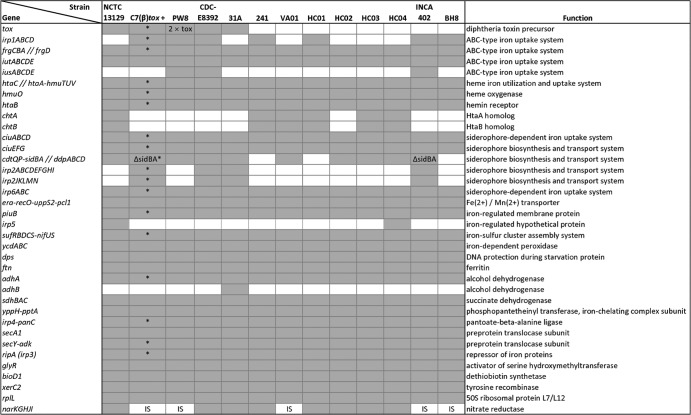
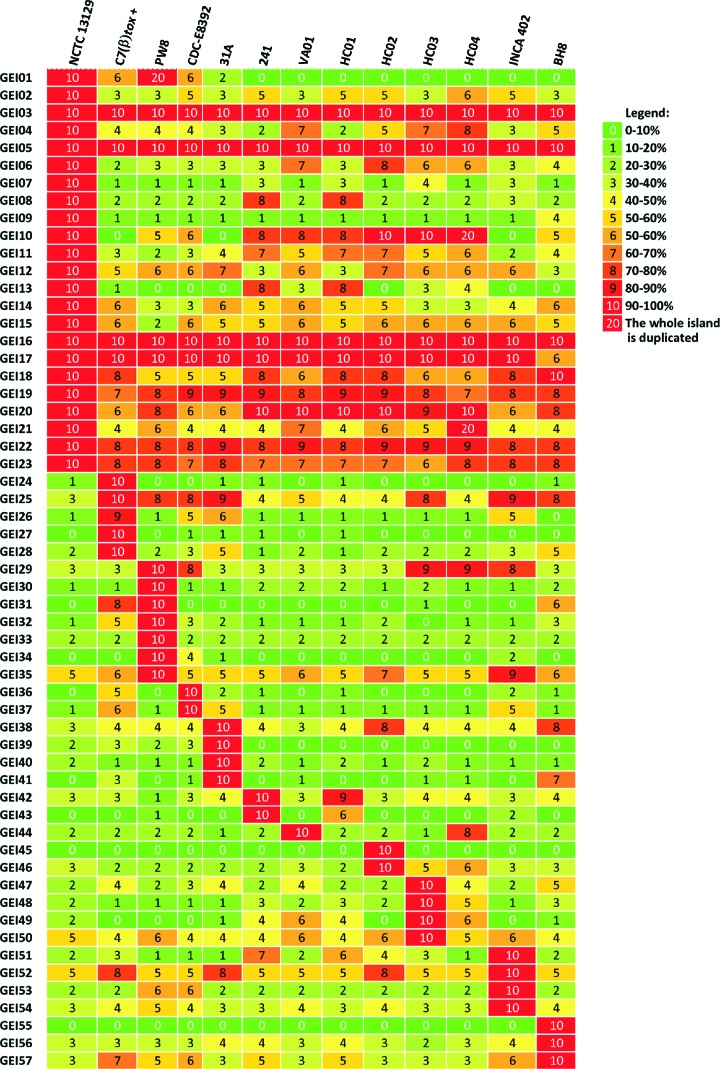
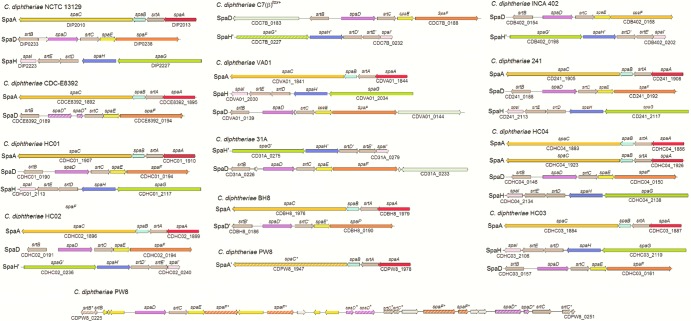
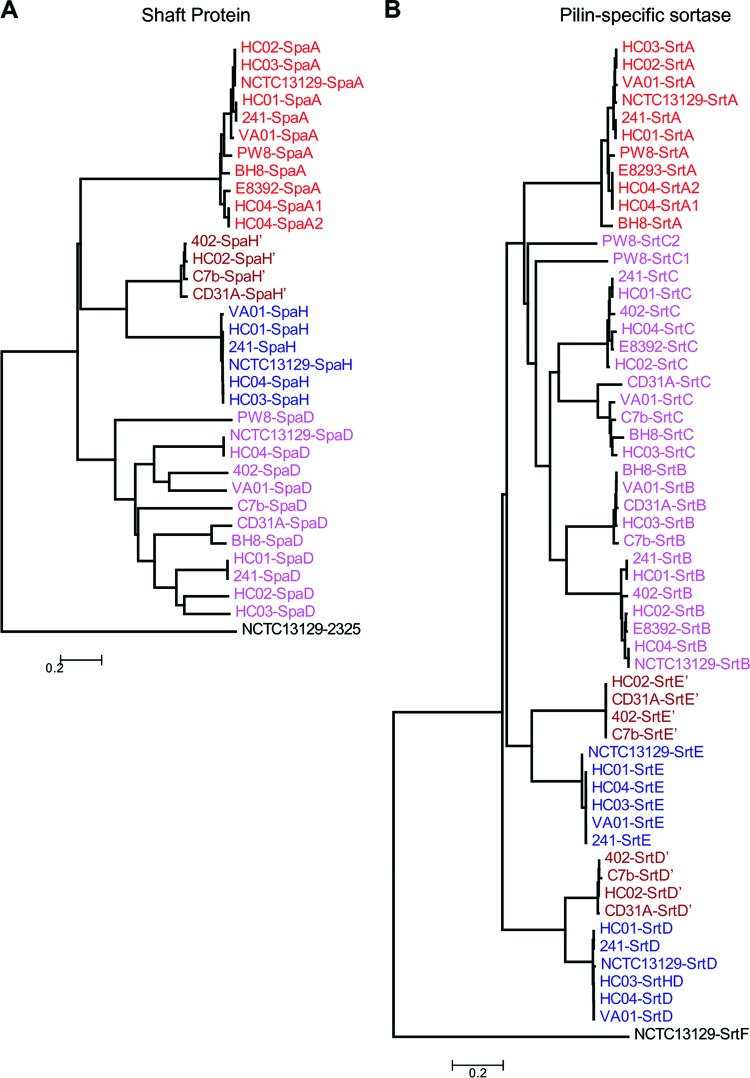
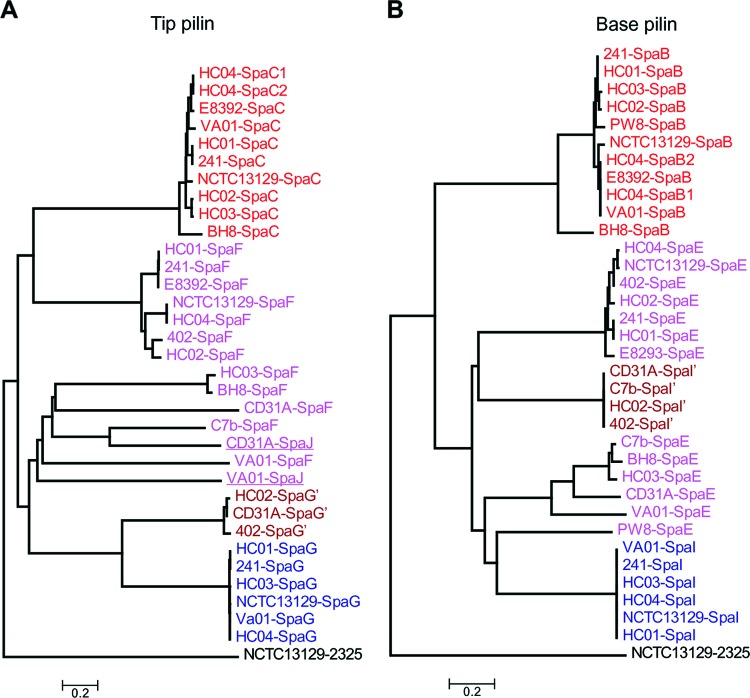
Corynebacterium diphtheriae is one of the most prominent human pathogens and the causative agent of the communicable disease diphtheria. The genomes of 12 strains isolated from patients with classical diphtheria, endocarditis, and pneumonia were completely sequenced and annotated. Including the genome of C. diphtheriae NCTC 13129, we herewith present a comprehensive comparative analysis of 13 strains and the first characterization of the pangenome of the species C. diphtheriae. Comparative genomics showed extensive synteny and revealed a core genome consisting of 1,632 conserved genes. The pangenome currently comprises 4,786 protein-coding regions and increases at an average of 65 unique genes per newly sequenced strain. Analysis of prophages carrying the diphtheria toxin gene tox revealed that the toxoid vaccine producer C. diphtheriae Park-Williams no. 8 has been lysogenized by two copies of the ω(tox)(+) phage, whereas C. diphtheriae 31A harbors a hitherto-unknown tox(+) corynephage. DNA binding sites of the tox-controlling regulator DtxR were detected by genome-wide motif searches. Comparative content analysis showed that the DtxR regulons exhibit marked differences due to gene gain, gene loss, partial gene deletion, and DtxR binding site depletion. Most predicted pathogenicity islands of C. diphtheriae revealed characteristics of horizontal gene transfer. The majority of these islands encode subunits of adhesive pili, which can play important roles in adhesion of C. diphtheriae to different host tissues. All sequenced isolates contain at least two pilus gene clusters. It appears that variation in the distributed genome is a common strategy of C. diphtheriae to establish differences in host-pathogen interactions.
Figures

References
-
- Andrews SC, Robinson AK, Rodriguez-Quinones F. 2003. Bacterial iron homeostasis. FEMS Microbiol. Rev. 27:215–237 - PubMed
-
- Badger JH, Olsen GJ. 1999. CRITICA: coding region identification tool invoking comparative analysis. Mol. Biol. Evol. 16:512–524 - PubMed
-
- Bottacini F, et al. 2010. Comparative genomics of the genus Bifidobacterium. Microbiology 156:3243–3254 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
