

Cell biology of spinocerebellar ataxia
- PMID: 22508507
- PMCID: PMC3328388
- DOI: 10.1083/jcb.201105092
Cell biology of spinocerebellar ataxia
Abstract
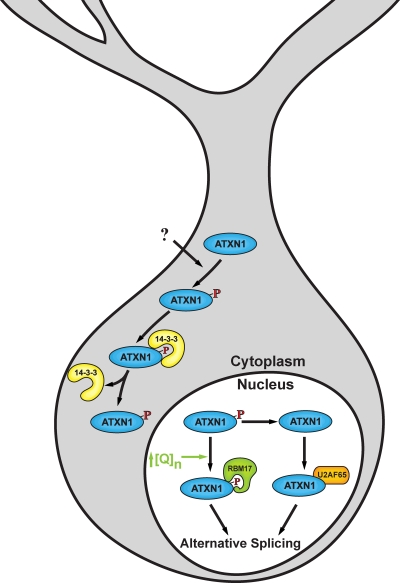
Ataxia is a neurological disorder characterized by loss of control of body movements. Spinocerebellar ataxia (SCA), previously known as autosomal dominant cerebellar ataxia, is a biologically robust group of close to 30 progressive neurodegenerative diseases. Six SCAs, including the more prevalent SCA1, SCA2, SCA3, and SCA6 along with SCA7 and SCA17 are caused by expansion of a CAG repeat that encodes a polyglutamine tract in the affected protein. How the mutated proteins in these polyglutamine SCAs cause disease is highly debated. Recent work suggests that the mutated protein contributes to pathogenesis within the context of its "normal" cellular function. Thus, understanding the cellular function of these proteins could aid in the development of therapeutics.
Figures


References
-
- Alves S., Nascimento-Ferreira I., Auregan G., Hassig R., Dufour N., Brouillet E., Pedroso de Lima M.C., Hantraye P., Pereira de Almeida L., Déglon N. 2008. Allele-specific RNA silencing of mutant ataxin-3 mediates neuroprotection in a rat model of Machado-Joseph disease. PLoS ONE. 3:e3341 10.1371/journal.pone.0003341 - DOI - PMC - PubMed
-
- Bichelmeier U., Schmidt T., Hübener J., Boy J., Rüttiger L., Häbig K., Poths S., Bonin M., Knipper M., Schmidt W.J., et al. 2007. Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: in vivo evidence. J. Neurosci. 27:7418–7428 10.1523/JNEUROSCI.4540-06.2007 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
