

Oxidative stress during mitochondrial biogenesis compromises mtDNA integrity in growing hearts and induces a global DNA repair response
- PMID: 22508755
- PMCID: PMC3413112
- DOI: 10.1093/nar/gks301
Oxidative stress during mitochondrial biogenesis compromises mtDNA integrity in growing hearts and induces a global DNA repair response
Abstract
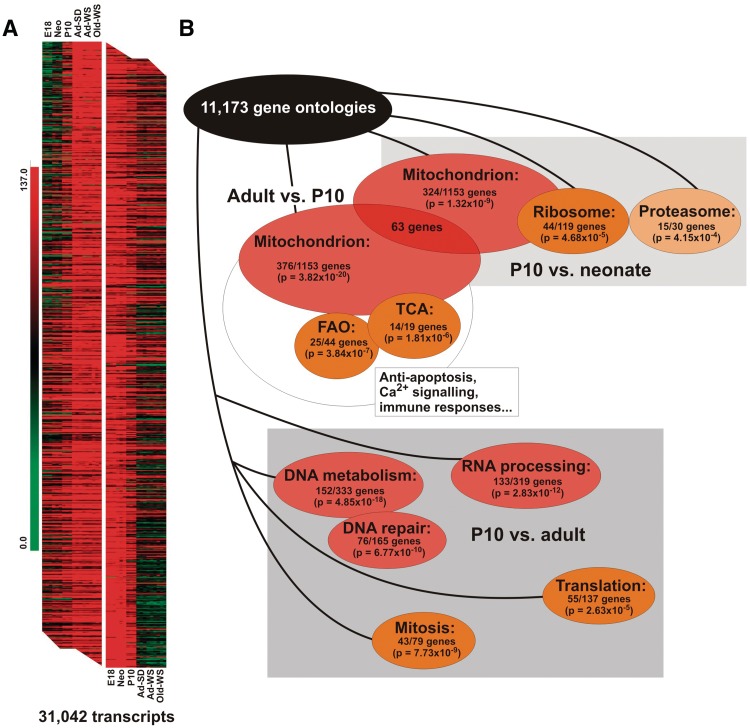
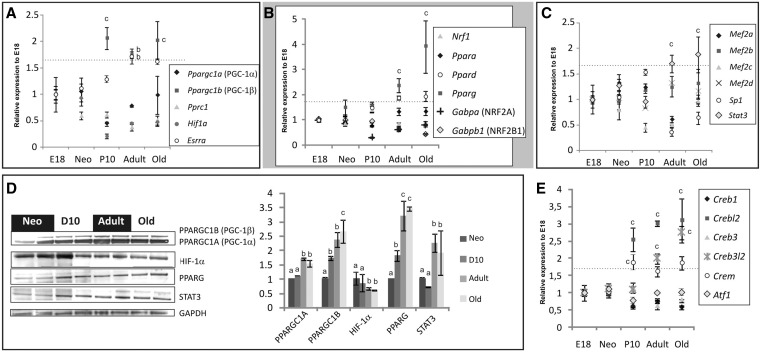
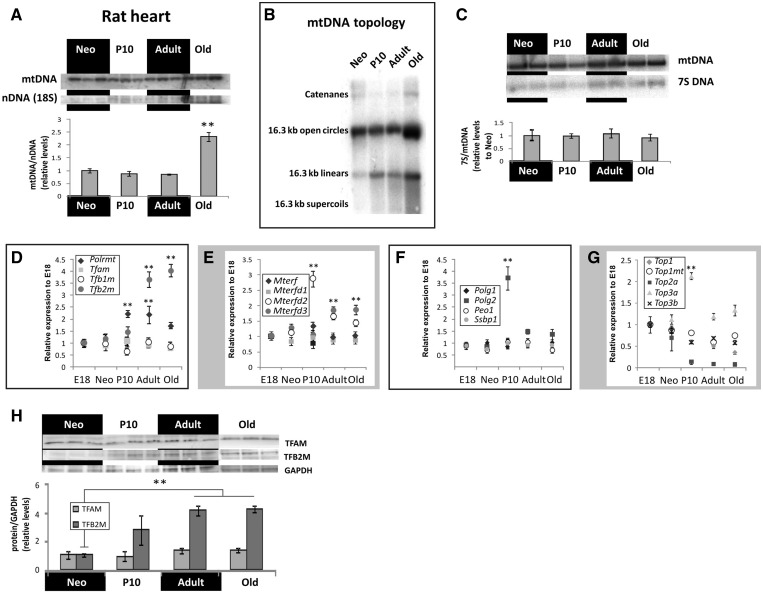
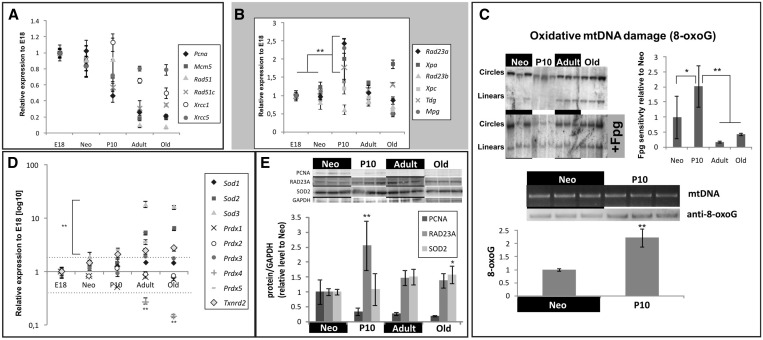
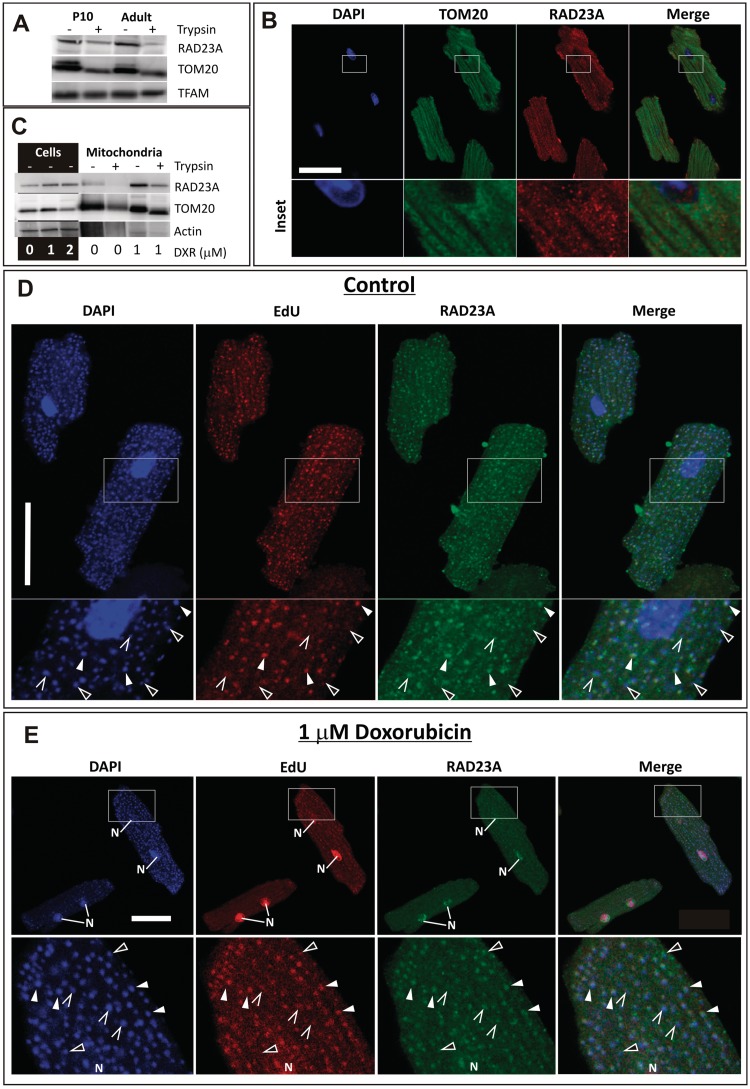
Cardiomyocyte development in mammals is characterized by a transition from hyperplastic to hypertrophic growth soon after birth. The rise of cardiomyocyte cell mass in postnatal life goes along with a proportionally bigger increase in the mitochondrial mass in response to growing energy requirements. Relatively little is known about the molecular processes regulating mitochondrial biogenesis and mitochondrial DNA (mtDNA) maintenance during developmental cardiac hypertrophy. Genome-wide transcriptional profiling revealed the activation of transcriptional regulatory circuits controlling mitochondrial biogenesis in growing rat hearts. In particular, we detected a specific upregulation of factors involved in mtDNA expression and translation. More surprisingly, we found a specific upregulation of DNA repair proteins directly linked to increased oxidative damage during heart mitochondrial biogenesis, but only relatively minor changes in the mtDNA replication machinery. Our study paves the way for improved understanding of mitochondrial biogenesis, mtDNA maintenance and physiological adaptation processes in the heart and provides the first evidence for the recruitment of nucleotide excision repair proteins to mtDNA in cardiomyocytes upon DNA damage.
Figures
Similar articles
-
Failed upregulation of TFAM protein and mitochondrial DNA in oxidatively deficient fibers of chronic obstructive pulmonary disease locomotor muscle.Skelet Muscle. 2016 Feb 18;6:10. doi: 10.1186/s13395-016-0083-9. eCollection 2016. Skelet Muscle. 2016. PMID: 26893822 Free PMC article.
-
Mitochondrial transcription factor A overexpression and base excision repair deficiency in the inner ear of rats with D-galactose-induced aging.FEBS J. 2011 Jul;278(14):2500-10. doi: 10.1111/j.1742-4658.2011.08176.x. Epub 2011 Jun 5. FEBS J. 2011. PMID: 21575134
-
Postnatal cardiomyocyte growth and mitochondrial reorganization cause multiple changes in the proteome of human cardiomyocytes.Mol Biosyst. 2013 Jun;9(6):1210-9. doi: 10.1039/c3mb25556e. Epub 2013 Mar 4. Mol Biosyst. 2013. PMID: 23459711
-
[Pathways for maintenance of mitochondrial DNA integrity and mitochondrial functions in cells exposed to ionizing radiation].Radiats Biol Radioecol. 2013 Mar-Apr;53(2):117-36. doi: 10.7868/s0869803113020045. Radiats Biol Radioecol. 2013. PMID: 23786028 Review. Russian.
-
The role of mitochondria in cardiac development and protection.Free Radic Biol Med. 2017 May;106:345-354. doi: 10.1016/j.freeradbiomed.2017.02.032. Epub 2017 Feb 17. Free Radic Biol Med. 2017. PMID: 28216385 Review.
Cited by
-
Mitochondrial Genetic and Epigenetic Regulations in Cancer: Therapeutic Potential.Int J Mol Sci. 2022 Jul 18;23(14):7897. doi: 10.3390/ijms23147897. Int J Mol Sci. 2022. PMID: 35887244 Free PMC article. Review.
-
Interaction of TNF with angiotensin II contributes to mitochondrial oxidative stress and cardiac damage in rats.PLoS One. 2012;7(10):e46568. doi: 10.1371/journal.pone.0046568. Epub 2012 Oct 9. PLoS One. 2012. PMID: 23056347 Free PMC article.
-
Drp1/Fis1-mediated mitochondrial fragmentation leads to lysosomal dysfunction in cardiac models of Huntington's disease.J Mol Cell Cardiol. 2019 Feb;127:125-133. doi: 10.1016/j.yjmcc.2018.12.004. Epub 2018 Dec 11. J Mol Cell Cardiol. 2019. PMID: 30550751 Free PMC article.
-
Metabolic modulation of mitochondrial mass during CD4+ T cell activation.Cell Chem Biol. 2023 Sep 21;30(9):1064-1075.e8. doi: 10.1016/j.chembiol.2023.08.008. Cell Chem Biol. 2023. PMID: 37716347 Free PMC article.
-
Differential Alterations of the Mitochondrial Morphology and Respiratory Chain Complexes during Postnatal Development of the Mouse Lung.Oxid Med Cell Longev. 2017;2017:9169146. doi: 10.1155/2017/9169146. Epub 2017 Dec 19. Oxid Med Cell Longev. 2017. PMID: 29430286 Free PMC article.
References
-
- Tam SK, Gu W, Mahdavi V, Nadal-Ginard B. Cardiac myocyte terminal differentiation. Potential for cardiac regeneration. Ann. N Y Acad. Sci. 1995;752:72–79. - PubMed
-
- Pasumarthi KB, Field LJ. Cardiomyocyte cell cycle regulation. Circ. Res. 2002;90:1044–1054. - PubMed
-
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006;7:589–600. - PubMed
-
- Goffart S, von Kleist-Retzow JC, Wiesner RJ. Regulation of mitochondrial proliferation in the heart: power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc. Res. 2004;64:198–207. - PubMed
