

Lysocardiolipin acyltransferase 1 (ALCAT1) controls mitochondrial DNA fidelity and biogenesis through modulation of MFN2 expression
- PMID: 22509026
- PMCID: PMC3345005
- DOI: 10.1073/pnas.1120043109
Lysocardiolipin acyltransferase 1 (ALCAT1) controls mitochondrial DNA fidelity and biogenesis through modulation of MFN2 expression
Abstract
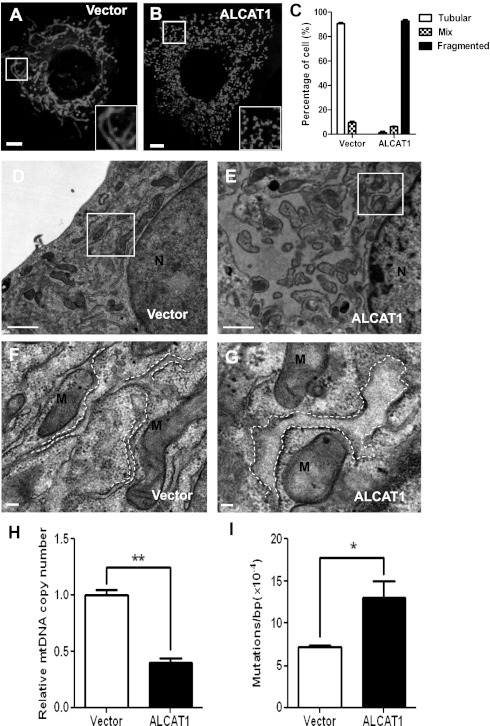
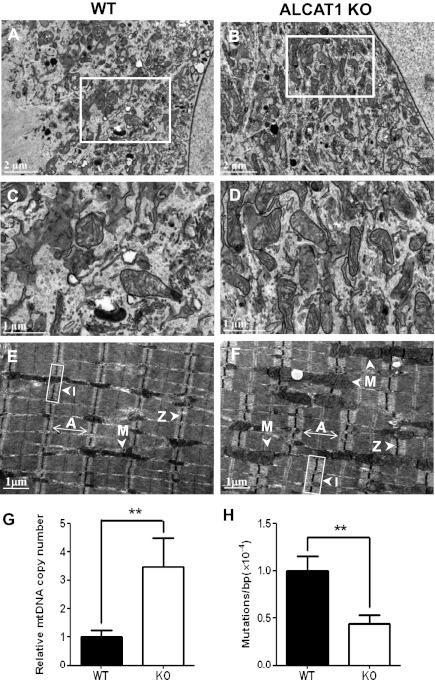
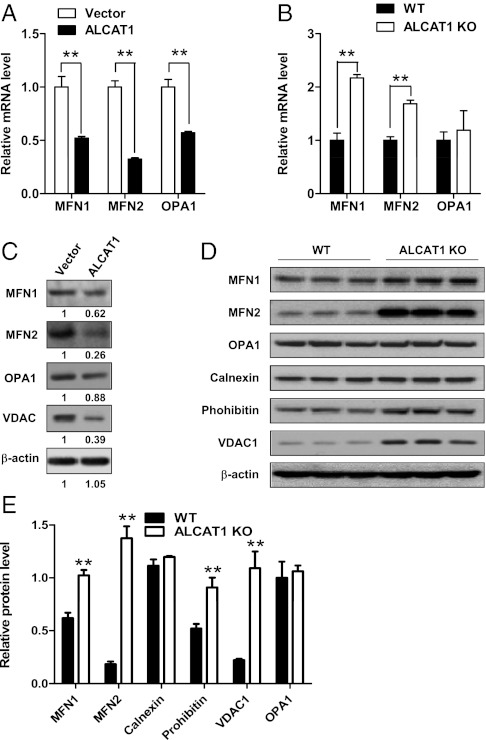
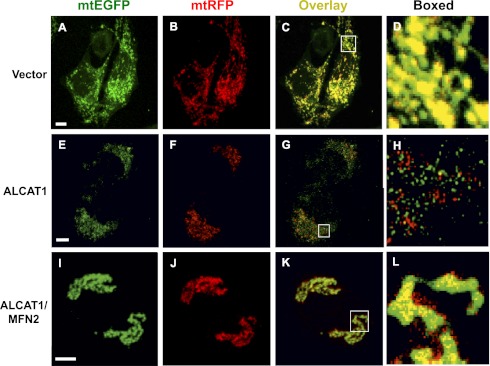
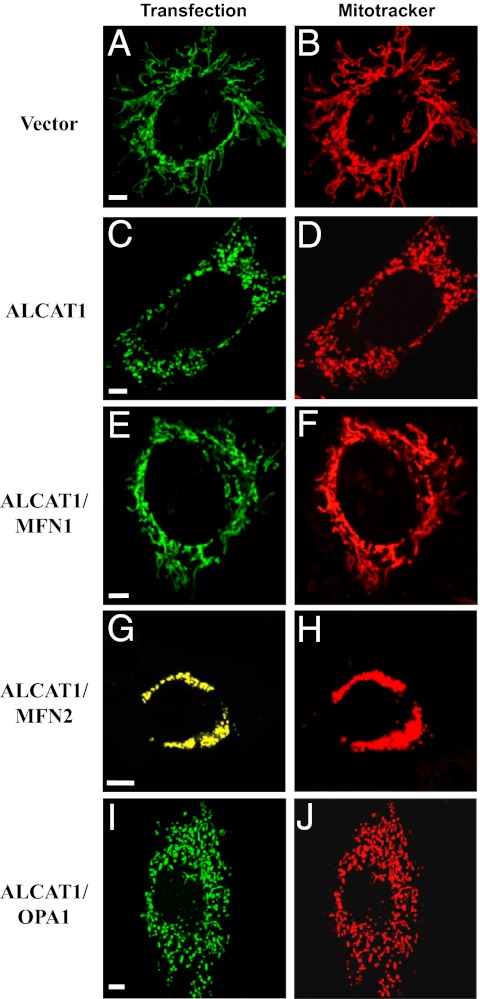
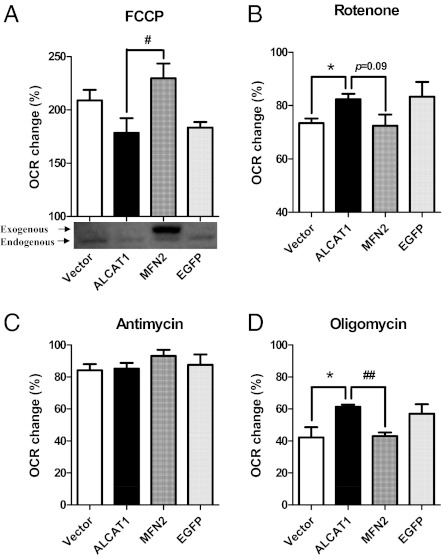
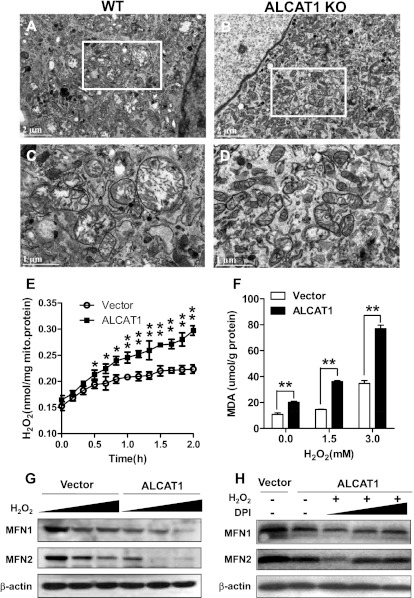
Oxidative stress causes mitochondrial fragmentation and dysfunction in age-related diseases through unknown mechanisms. Cardiolipin (CL) is a phospholipid required for mitochondrial oxidative phosphorylation. The function of CL is determined by its acyl composition, which is significantly altered by the onset of age-related diseases. Here, we examine a role of acyl-CoA:lysocardiolipin acyltransferase lysocardiolipin acyltransferase 1 (ALCAT1), a lysocardiolipin acyltransferase that catalyzes pathological CL remodeling, in mitochondrial biogenesis. We show that overexpression of ALCAT1 causes mitochondrial fragmentation through oxidative stress and depletion of mitofusin mitofusin 2 (MFN2) expression. Strikingly, ALCAT1 overexpression also leads to mtDNA instability and depletion that are reminiscent of MFN2 deficiency. Accordingly, expression of MFN2 completely rescues mitochondrial fusion defect and respiratory dysfunction. Furthermore, ablation of ALCAT1 prevents mitochondrial fragmentation from oxidative stress by up-regulating MFN2 expression, mtDNA copy number, and mtDNA fidelity. Together, these findings reveal an unexpected role of CL remodeling in mitochondrial biogenesis, linking oxidative stress by ALCAT1 to mitochondrial fusion defect.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
