

Cryptic distant relatives are common in both isolated and cosmopolitan genetic samples
- PMID: 22509285
- PMCID: PMC3317976
- DOI: 10.1371/journal.pone.0034267
Cryptic distant relatives are common in both isolated and cosmopolitan genetic samples
Abstract
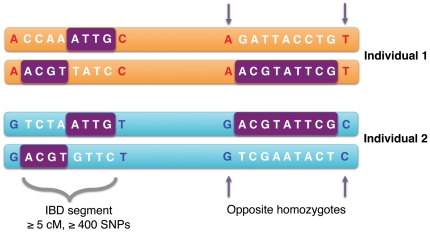
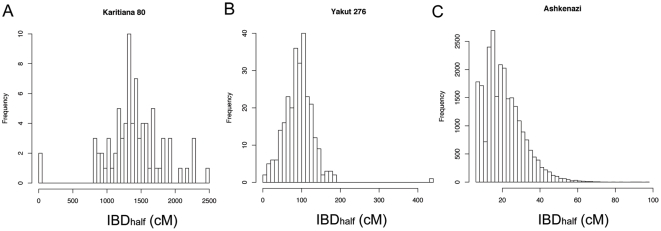
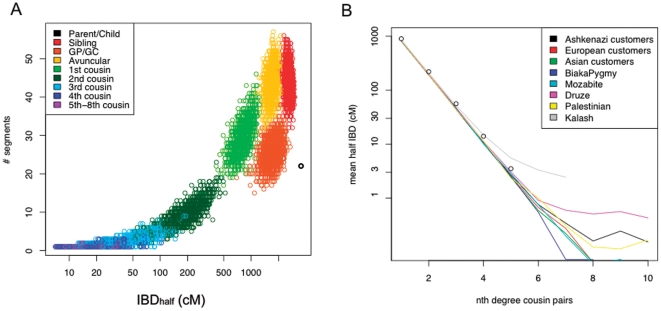
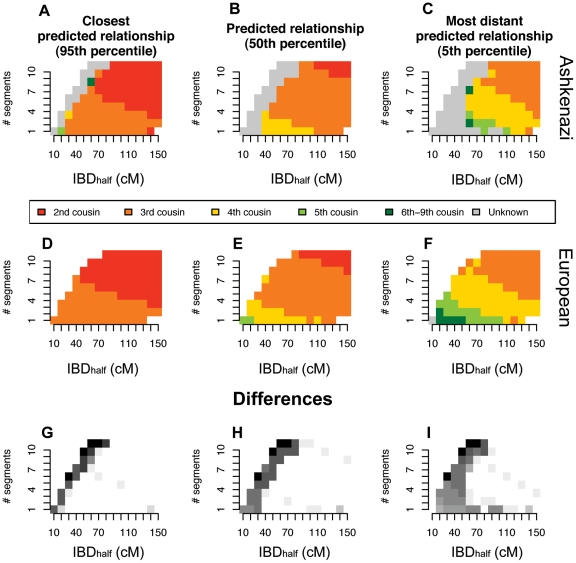
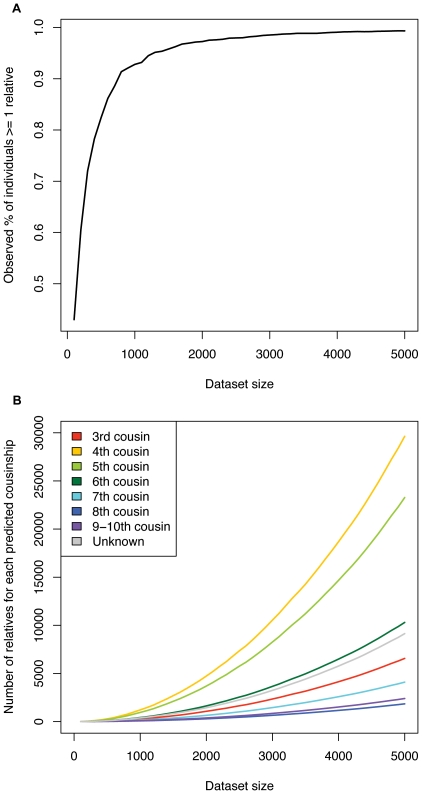
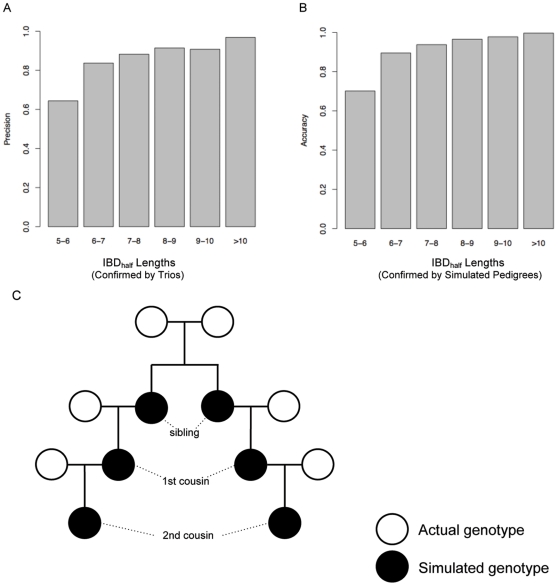
Although a few hundred single nucleotide polymorphisms (SNPs) suffice to infer close familial relationships, high density genome-wide SNP data make possible the inference of more distant relationships such as 2(nd) to 9(th) cousinships. In order to characterize the relationship between genetic similarity and degree of kinship given a timeframe of 100-300 years, we analyzed the sharing of DNA inferred to be identical by descent (IBD) in a subset of individuals from the 23andMe customer database (n = 22,757) and from the Human Genome Diversity Panel (HGDP-CEPH, n = 952). With data from 121 populations, we show that the average amount of DNA shared IBD in most ethnolinguistically-defined populations, for example Native American groups, Finns and Ashkenazi Jews, differs from continentally-defined populations by several orders of magnitude. Via extensive pedigree-based simulations, we determined bounds for predicted degrees of relationship given the amount of genomic IBD sharing in both endogamous and 'unrelated' population samples. Using these bounds as a guide, we detected tens of thousands of 2(nd) to 9(th) degree cousin pairs within a heterogenous set of 5,000 Europeans. The ubiquity of distant relatives, detected via IBD segments, in both ethnolinguistic populations and in large 'unrelated' populations samples has important implications for genetic genealogy, forensics and genotype/phenotype mapping studies.
Conflict of interest statement
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
