

Domain enhanced lookup time accelerated BLAST
- PMID: 22510480
- PMCID: PMC3438057
- DOI: 10.1186/1745-6150-7-12
Domain enhanced lookup time accelerated BLAST
Abstract
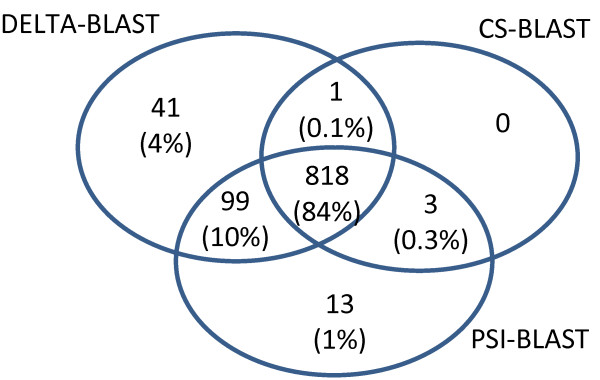
Background: BLAST is a commonly-used software package for comparing a query sequence to a database of known sequences; in this study, we focus on protein sequences. Position-specific-iterated BLAST (PSI-BLAST) iteratively searches a protein sequence database, using the matches in round i to construct a position-specific score matrix (PSSM) for searching the database in round i + 1. Biegert and Söding developed Context-sensitive BLAST (CS-BLAST), which combines information from searching the sequence database with information derived from a library of short protein profiles to achieve better homology detection than PSI-BLAST, which builds its PSSMs from scratch.
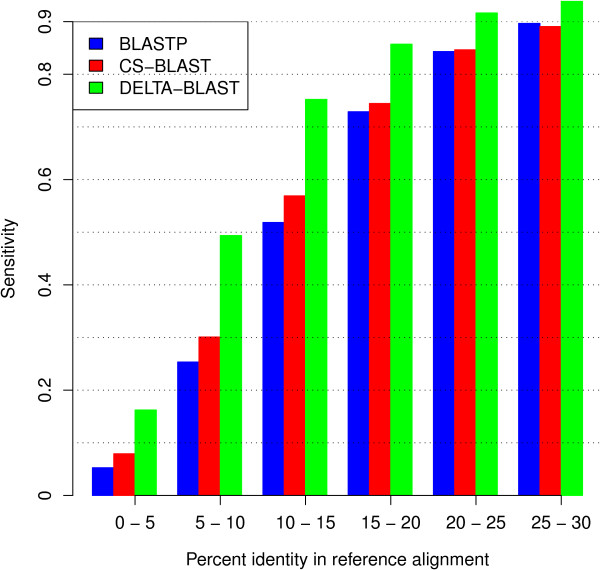
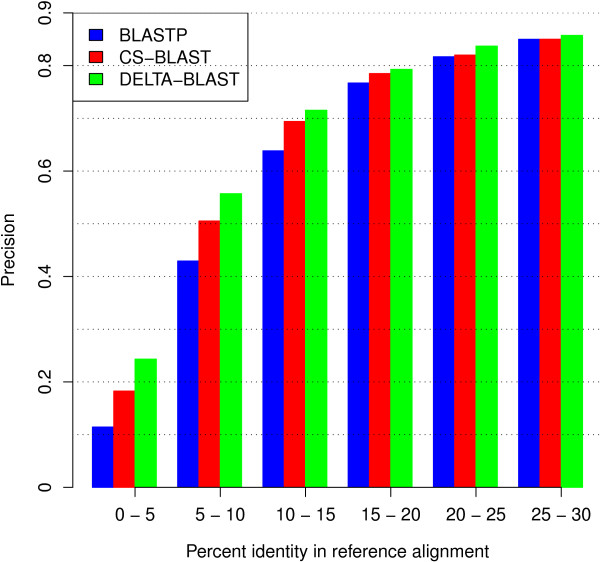
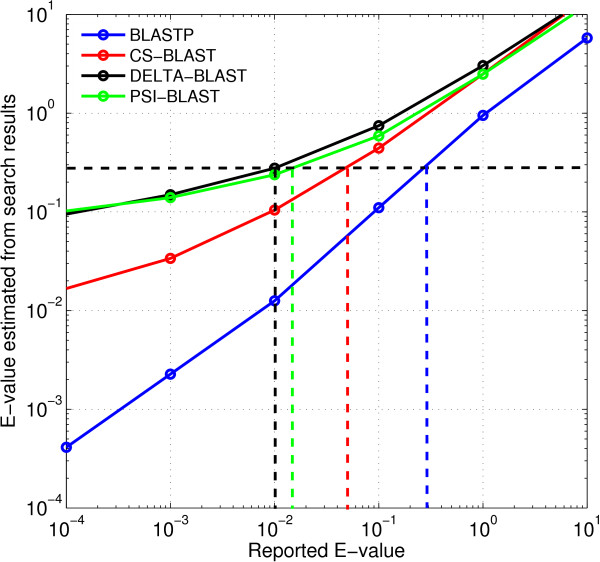
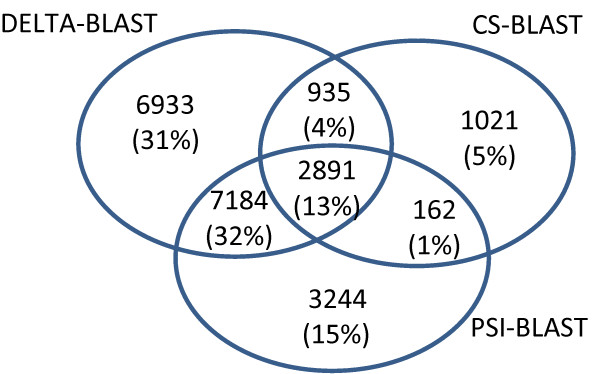
Results: We describe a new method, called domain enhanced lookup time accelerated BLAST (DELTA-BLAST), which searches a database of pre-constructed PSSMs before searching a protein-sequence database, to yield better homology detection. For its PSSMs, DELTA-BLAST employs a subset of NCBI's Conserved Domain Database (CDD). On a test set derived from ASTRAL, with one round of searching, DELTA-BLAST achieves a ROC5000 of 0.270 vs. 0.116 for CS-BLAST. The performance advantage diminishes in iterated searches, but DELTA-BLAST continues to achieve better ROC scores than CS-BLAST.
Conclusions: DELTA-BLAST is a useful program for the detection of remote protein homologs. It is available under the "Protein BLAST" link at http://blast.ncbi.nlm.nih.gov.
Figures






References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
