

Regulation of NMDA receptor transport: a KIF17-cargo binding/releasing underlies synaptic plasticity and memory in vivo
- PMID: 22514311
- PMCID: PMC6703487
- DOI: 10.1523/JNEUROSCI.0718-12.2012
Regulation of NMDA receptor transport: a KIF17-cargo binding/releasing underlies synaptic plasticity and memory in vivo
Abstract
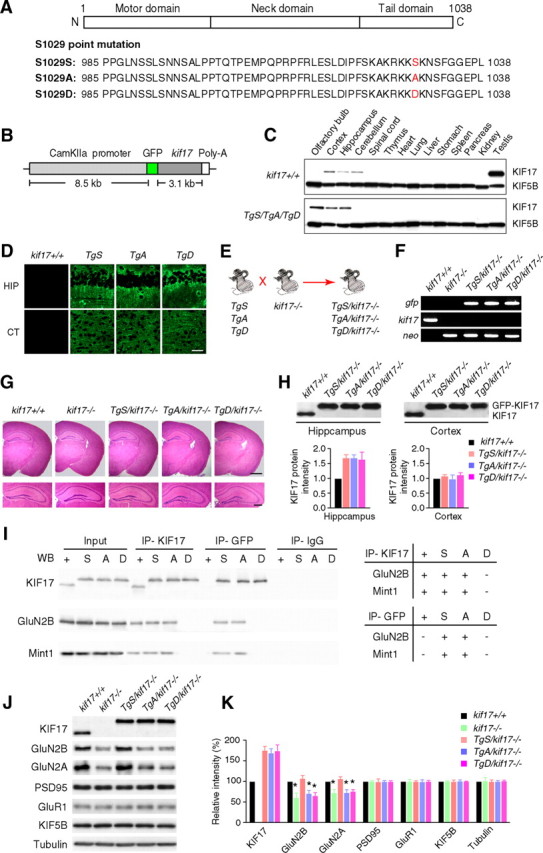
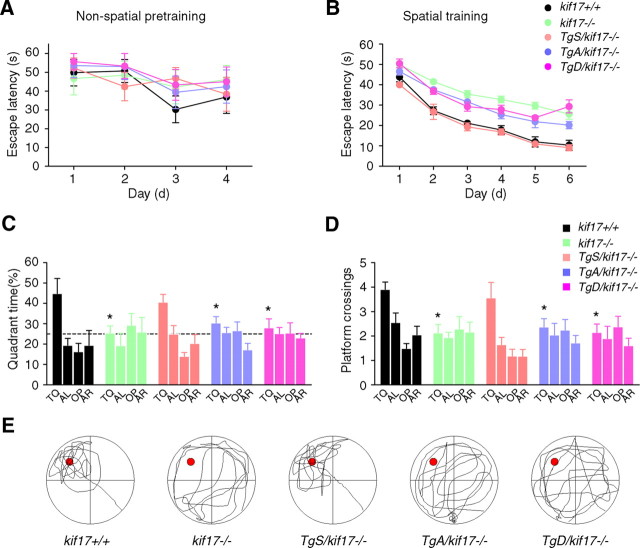
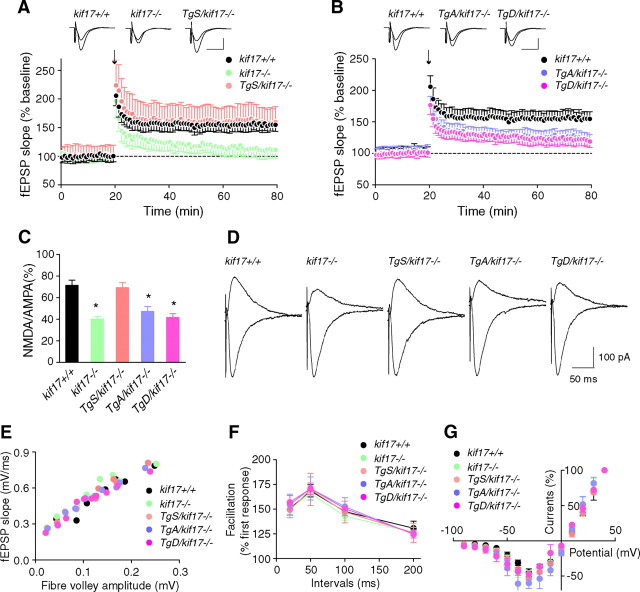
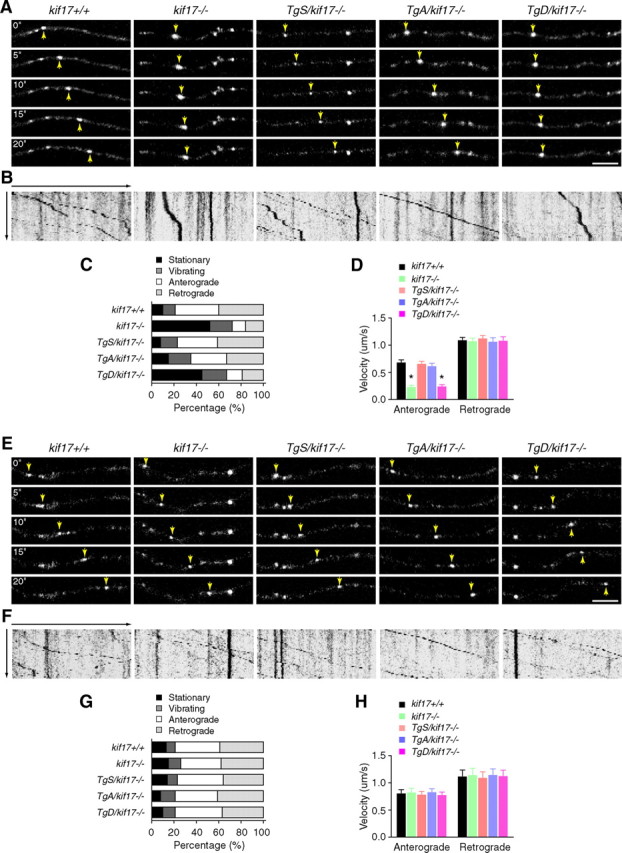
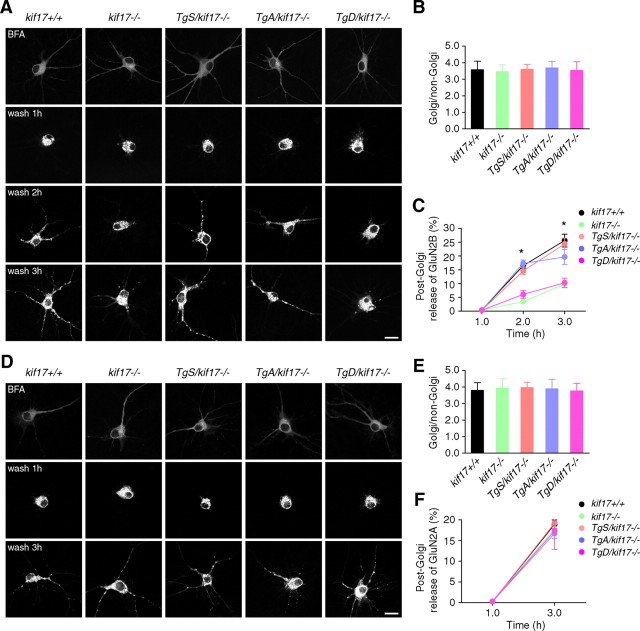
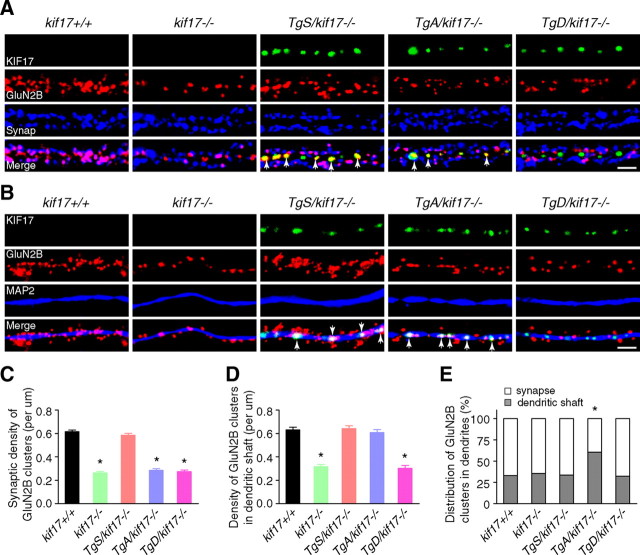
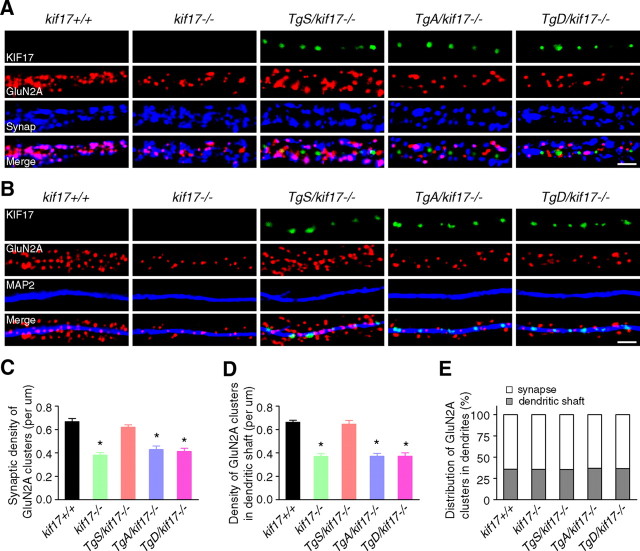
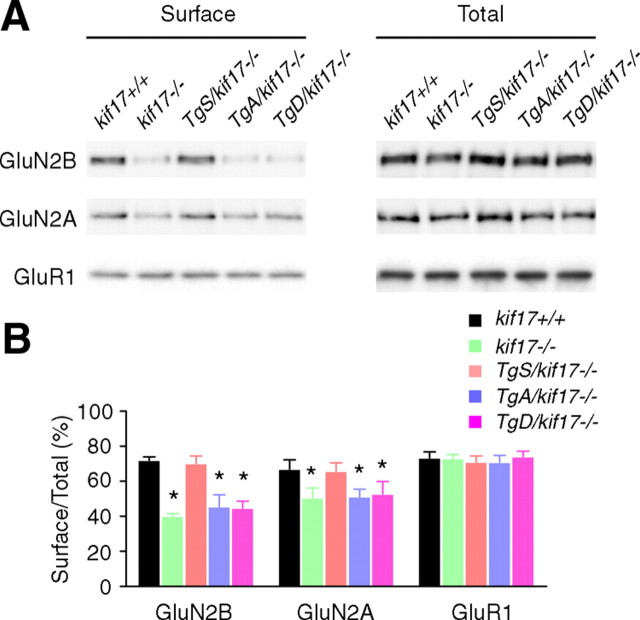
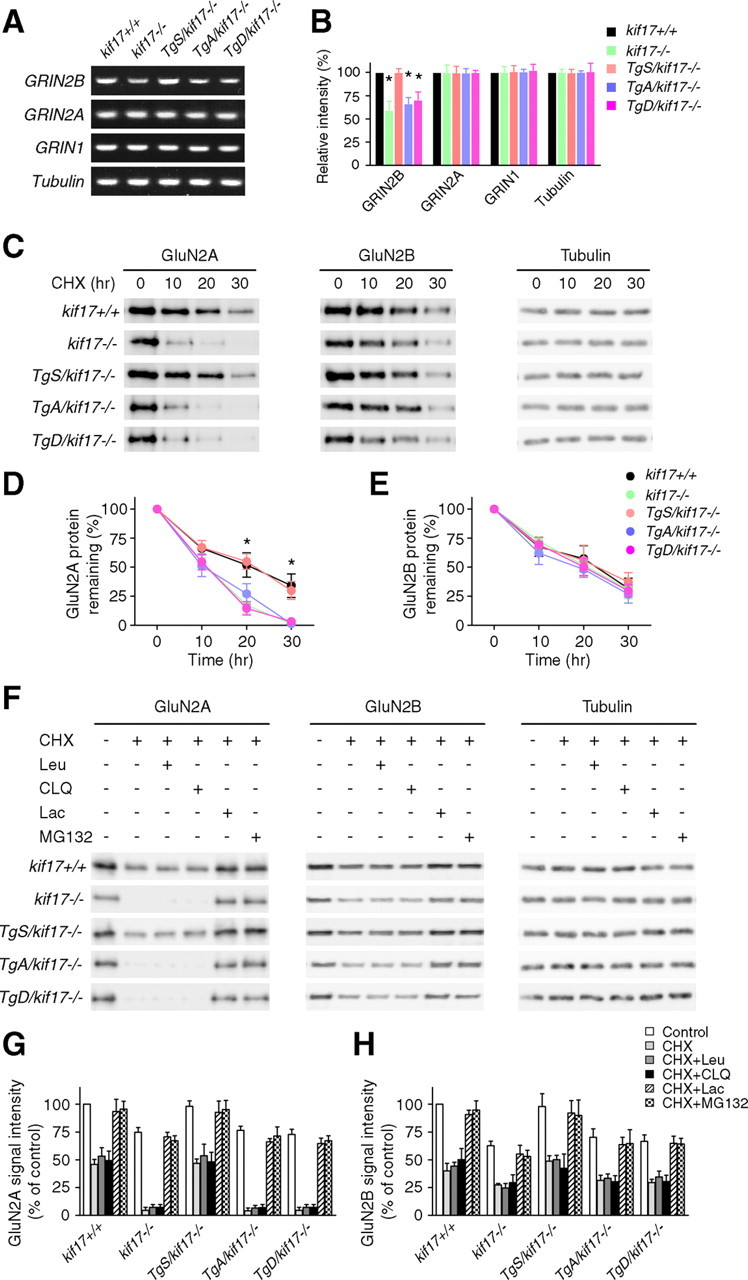
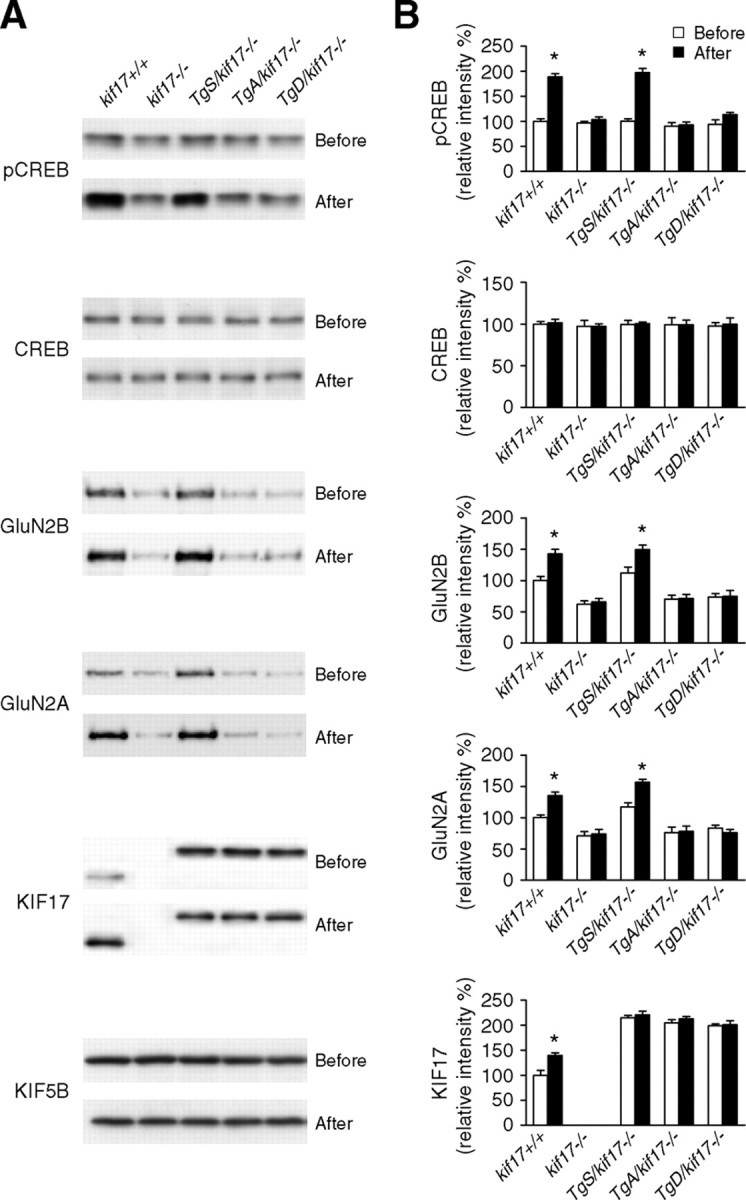
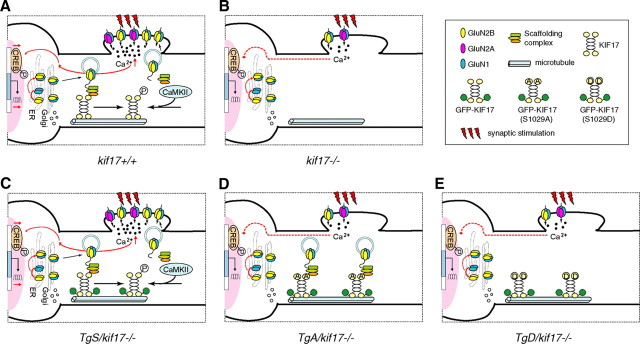
Regulation of NMDA receptor trafficking is crucial to modulate neuronal communication. Ca(2+)/calmodulin-dependent protein kinase phosphorylates the tail domain of KIF17, a member of the kinesin superfamily, to control NMDA receptor subunit 2B (GluN2B) transport by changing the KIF17-cargo interaction in vitro. However, the mechanisms of regulation of GluN2B transport in vivo and its physiological significance are unknown. We generated transgenic mice carrying wild-type KIF17 (TgS), or KIF17 with S1029A (TgA) or S1029D (TgD) phosphomimic mutations in kif17(-/-) background. TgA/kif17(-/-) and TgD/kif17(-/-) mice exhibited reductions in synaptic NMDA receptors because of their inability to load/unload GluN2B onto/from KIF17, leading to impaired neuronal plasticity, CREB activation, and spatial memory. Expression of GFP-KIF17 in TgS/kif17(-/-) mouse neurons rescued the synaptic and behavioral defects of kif17(-/-) mice. These results suggest that phosphorylation-based regulation of NMDA receptor transport is critical for learning and memory in vivo.
Figures
Similar articles
-
Molecular motor KIF17 is fundamental for memory and learning via differential support of synaptic NR2A/2B levels.Neuron. 2011 Apr 28;70(2):310-25. doi: 10.1016/j.neuron.2011.02.049. Neuron. 2011. PMID: 21521616
-
TrkB phosphorylation by Cdk5 is required for activity-dependent structural plasticity and spatial memory.Nat Neurosci. 2012 Nov;15(11):1506-15. doi: 10.1038/nn.3237. Epub 2012 Oct 14. Nat Neurosci. 2012. PMID: 23064382 Free PMC article.
-
AKAP150-anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+-permeable AMPA receptors.J Neurosci. 2012 Oct 24;32(43):15036-52. doi: 10.1523/JNEUROSCI.3326-12.2012. J Neurosci. 2012. PMID: 23100425 Free PMC article.
-
Protein-protein interactions at the NMDA receptor complex: From synaptic retention to synaptonuclear protein messengers.Neuropharmacology. 2021 Jun 1;190:108551. doi: 10.1016/j.neuropharm.2021.108551. Epub 2021 Apr 2. Neuropharmacology. 2021. PMID: 33819458 Review.
-
Hippocampal synaptic plasticity, spatial memory and anxiety.Nat Rev Neurosci. 2014 Mar;15(3):181-92. doi: 10.1038/nrn3677. Nat Rev Neurosci. 2014. PMID: 24552786 Review.
Cited by
-
R869C mutation in molecular motor KIF17 gene is involved in dementia with Lewy bodies.Alzheimers Dement (Amst). 2021 Jun 4;13(1):e12143. doi: 10.1002/dad2.12143. eCollection 2021. Alzheimers Dement (Amst). 2021. PMID: 34124335 Free PMC article.
-
An activity-dependent local transport regulation via degradation and synthesis of KIF17 underlying cognitive flexibility.Sci Adv. 2020 Dec 16;6(51):eabc8355. doi: 10.1126/sciadv.abc8355. Print 2020 Dec. Sci Adv. 2020. PMID: 33328231 Free PMC article.
-
Dual and opposing roles for the kinesin-2 motor, KIF17, in Hedgehog-dependent cerebellar development.Sci Adv. 2024 Apr 26;10(17):eade1650. doi: 10.1126/sciadv.ade1650. Epub 2024 Apr 26. Sci Adv. 2024. PMID: 38669326 Free PMC article.
-
Beta-arrestin 1 regulation of reward-motivated behaviors and glutamatergic function.PLoS One. 2017 Oct 3;12(10):e0185796. doi: 10.1371/journal.pone.0185796. eCollection 2017. PLoS One. 2017. PMID: 28973019 Free PMC article.
-
Role of tubulin post-translational modifications in peripheral neuropathy.Exp Neurol. 2023 Feb;360:114274. doi: 10.1016/j.expneurol.2022.114274. Epub 2022 Nov 13. Exp Neurol. 2023. PMID: 36379274 Free PMC article. Review.
References
-
- Arimura N, Kaibuchi K. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat Rev Neurosci. 2007;8:194–205. - PubMed
-
- Arimura N, Kimura T, Nakamuta S, Taya S, Funahashi Y, Hattori A, Shimada A, Ménager C, Kawabata S, Fujii K, Iwamatsu A, Segal RA, Fukuda M, Kaibuchi K. Anterograde transport of TrkB in axons is mediated by direct interaction with Slp1 and Rab27. Dev Cell. 2009;16:675–686. - PubMed
-
- Bannerman DM, Good MA, Butcher SP, Ramsay M, Morris RGM. Distinct components of spatial learning revealed by prior training and NMDA receptor blockade. Nature. 1995;378:182–186. - PubMed
-
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–353. - PubMed
-
- Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous