

Analysis of a Streptococcus pyogenes puerperal sepsis cluster by use of whole-genome sequencing
- PMID: 22518858
- PMCID: PMC3405596
- DOI: 10.1128/JCM.00675-12
Analysis of a Streptococcus pyogenes puerperal sepsis cluster by use of whole-genome sequencing
Abstract
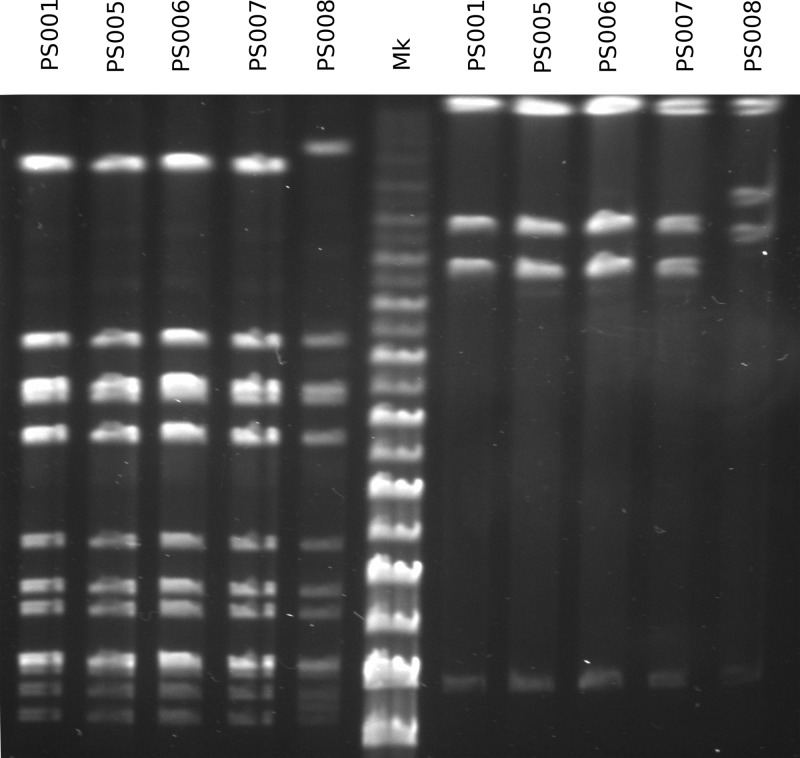
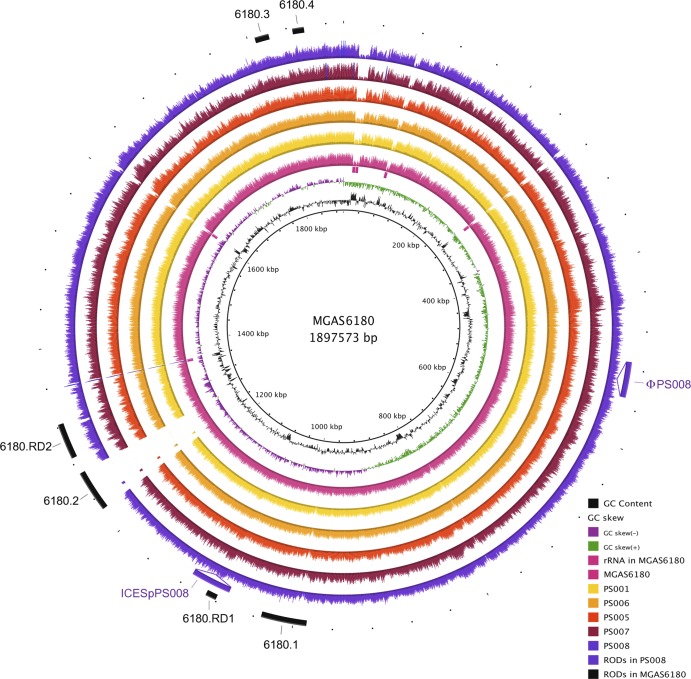
Between June and November 2010, a concerning rise in the number of cases of puerperal sepsis, a postpartum pelvic bacterial infection contracted by women after childbirth, was observed in the New South Wales, Australia, hospital system. Group A streptococcus (GAS; Streptococcus pyogenes) isolates PS001 to PS011 were recovered from nine patients. Pulsed-field gel electrophoresis and emm sequence typing revealed that GAS of emm1.40, emm75.0, emm77.0, emm89.0, and emm89.9 were each recovered from a single patient, ruling out a single source of infection. However, emm28.8 GAS were recovered from four different patients. To investigate the relatedness of these emm28 isolates, whole-genome sequencing was undertaken and the genome sequences were compared to the genome sequence of the emm28.4 reference strain, MGAS6180. A total of 186 single nucleotide polymorphisms were identified, for which the phylogenetic reconstruction indicated an outbreak of a polyclonal nature. While two isolates collected from different hospitals were not closely related, isolates from two puerperal sepsis patients from the same hospital were indistinguishable, suggesting patient-to-patient transmission or infection from a common source. The results of this study indicate that traditional typing protocols, such as pulsed-field gel electrophoresis, may not be sensitive enough to allow fine epidemiological discrimination of closely related bacterial isolates. Whole-genome sequencing presents a valid alternative that allows accurate fine-scale epidemiological investigation of bacterial infectious disease.
Figures

References
-
- Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. 2011. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402 doi:10.1186/1471-2164-12-402 - DOI - PMC - PubMed
-
- Aziz RK, et al. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75 doi:10.1186/1471-2164-9-75 - DOI - PMC - PubMed
-
- Aziz RK, Nizet V. 2010. Pathogen microevolution in high resolution. Sci. Transl. Med. 2:16ps4 - PubMed
-
- Beres SB, Musser JM. 2007. Contribution of exogenous genetic elements to the group A Streptococcus metagenome. PLoS One 2:e800 doi:10.1371/journal.pone.0000800 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
