

Inborn errors of human JAKs and STATs
- PMID: 22520845
- PMCID: PMC3334867
- DOI: 10.1016/j.immuni.2012.03.016
Inborn errors of human JAKs and STATs
Abstract
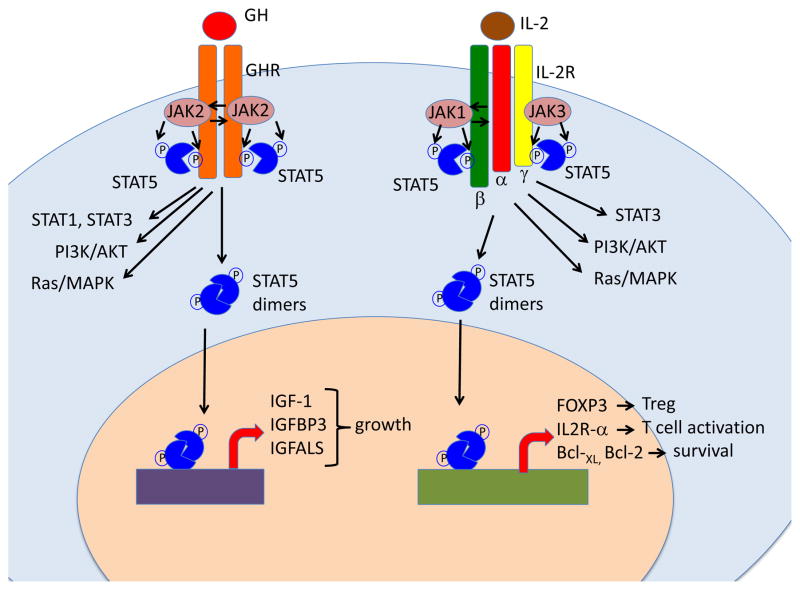
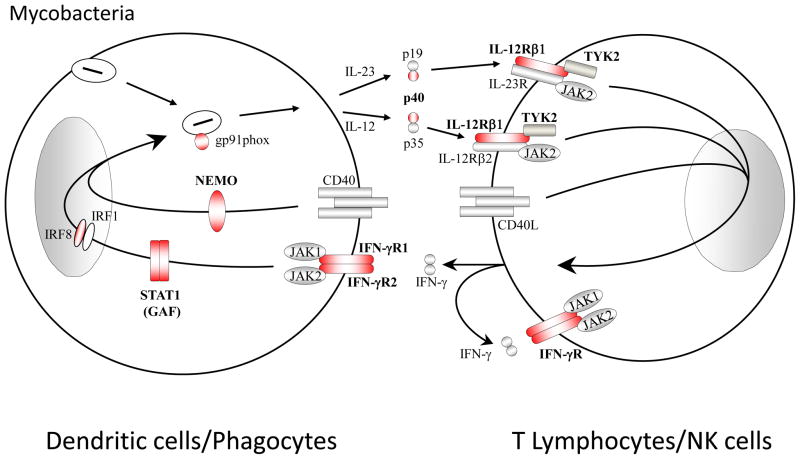
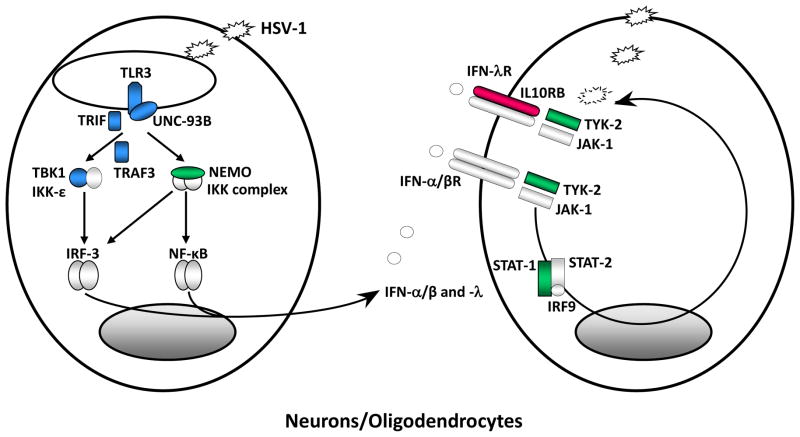
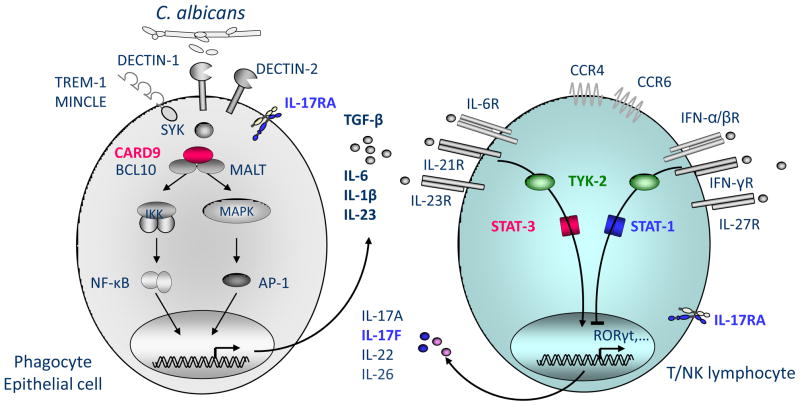
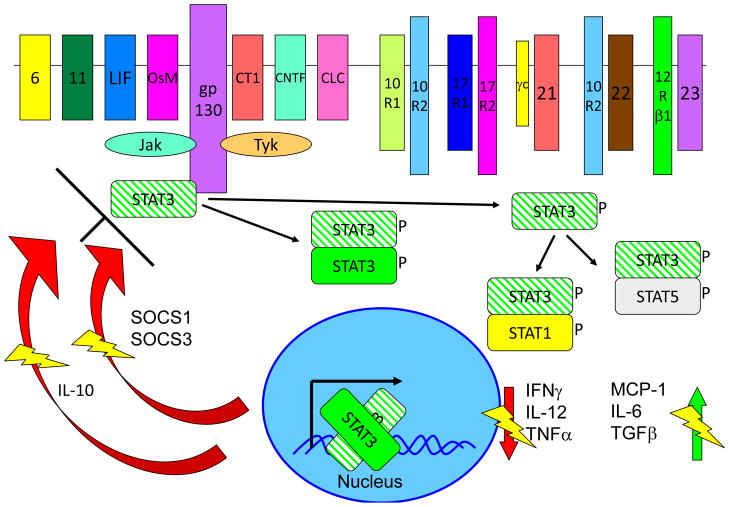
Inborn errors of the genes encoding two of the four human JAKs (JAK3 and TYK2) and three of the six human STATs (STAT1, STAT3, and STAT5B) have been described. We review the disorders arising from mutations in these five genes, highlighting the way in which the molecular and cellular pathogenesis of these conditions has been clarified by the discovery of inborn errors of cytokines, hormones, and their receptors, including those interacting with JAKs and STATs. The phenotypic similarities between mice and humans lacking individual JAK-STAT components suggest that the functions of JAKs and STATs are largely conserved in mammals. However, a wide array of phenotypic differences has emerged between mice and humans carrying biallelic null alleles of JAK3, TYK2, STAT1, or STAT5B. Moreover, the high degree of allelic heterogeneity at the human JAK3, TYK2, STAT1, and STAT3 loci has revealed highly diverse immunological and clinical phenotypes, which had not been anticipated.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Farrar JD, et al. Selective loss of type I interferon-induced STAT4 activation caused by a minisatellite insertion in mouse Stat2. Nat Immunol. 2000;1:65–69. - PubMed
-
- Casanova JL, Abel L. The human model: a genetic dissection of immunity to infection in natural conditions. Nat Rev Immunol. 2004;4:55–66. - PubMed
-
- Glanzmann E, Riniker P. [Essential lymphocytophthisis; new clinical aspect of infant pathology] Annales paediatrici International review of pediatrics. 1950;175:1–32. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
