

lobSTR: A short tandem repeat profiler for personal genomes
- PMID: 22522390
- PMCID: PMC3371701
- DOI: 10.1101/gr.135780.111
lobSTR: A short tandem repeat profiler for personal genomes
Abstract
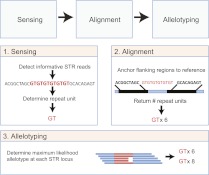
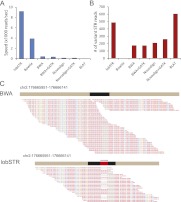
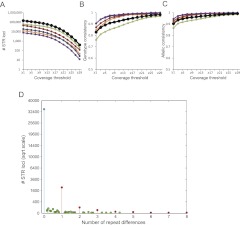
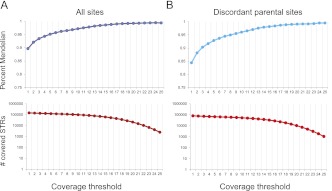
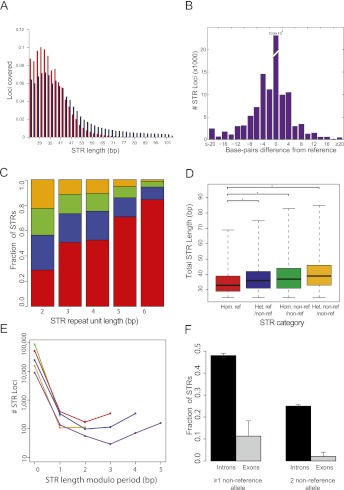
Short tandem repeats (STRs) have a wide range of applications, including medical genetics, forensics, and genetic genealogy. High-throughput sequencing (HTS) has the potential to profile hundreds of thousands of STR loci. However, mainstream bioinformatics pipelines are inadequate for the task. These pipelines treat STR mapping as gapped alignment, which results in cumbersome processing times and a biased sampling of STR alleles. Here, we present lobSTR, a novel method for profiling STRs in personal genomes. lobSTR harnesses concepts from signal processing and statistical learning to avoid gapped alignment and to address the specific noise patterns in STR calling. The speed and reliability of lobSTR exceed the performance of current mainstream algorithms for STR profiling. We validated lobSTR's accuracy by measuring its consistency in calling STRs from whole-genome sequencing of two biological replicates from the same individual, by tracing Mendelian inheritance patterns in STR alleles in whole-genome sequencing of a HapMap trio, and by comparing lobSTR results to traditional molecular techniques. Encouraged by the speed and accuracy of lobSTR, we used the algorithm to conduct a comprehensive survey of STR variations in a deeply sequenced personal genome. We traced the mutation dynamics of close to 100,000 STR loci and observed more than 50,000 STR variations in a single genome. lobSTR's implementation is an end-to-end solution. The package accepts raw sequencing reads and provides the user with the genotyping results. It is written in C/C++, includes multi-threading capabilities, and is compatible with the BAM format.
Figures
References
-
- Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM, Lafreniere RG, Rommens JM, Uyama E, Nohira O, et al. 1998. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet 18: 164–167 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous