

Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan
- PMID: 22522421
- PMCID: PMC3378661
- DOI: 10.1038/ng.2253
Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan
Abstract
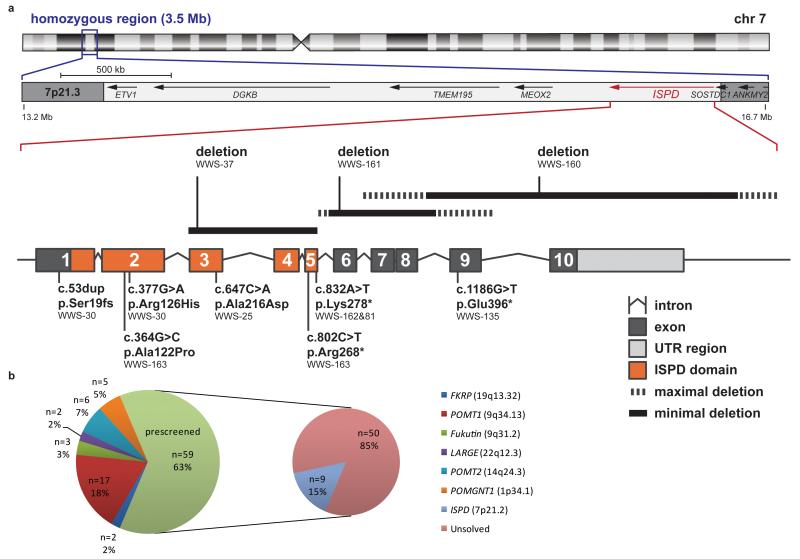
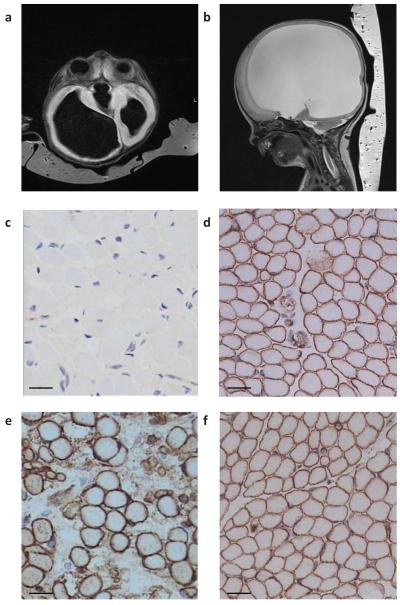
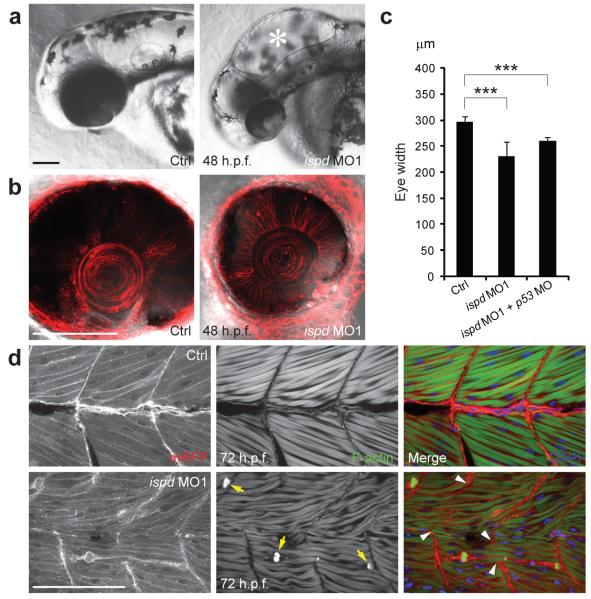
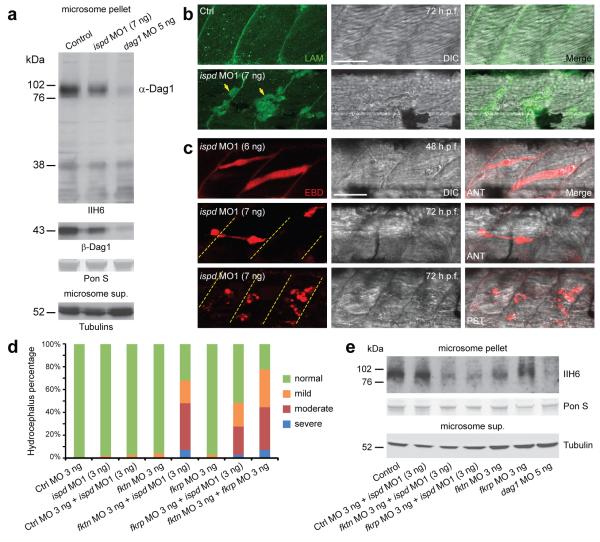
Walker-Warburg syndrome (WWS) is an autosomal recessive multisystem disorder characterized by complex eye and brain abnormalities with congenital muscular dystrophy (CMD) and aberrant a-dystroglycan glycosylation. Here we report mutations in the ISPD gene (encoding isoprenoid synthase domain containing) as the second most common cause of WWS. Bacterial IspD is a nucleotidyl transferase belonging to a large glycosyltransferase family, but the role of the orthologous protein in chordates is obscure to date, as this phylum does not have the corresponding non-mevalonate isoprenoid biosynthesis pathway. Knockdown of ispd in zebrafish recapitulates the human WWS phenotype with hydrocephalus, reduced eye size, muscle degeneration and hypoglycosylated a-dystroglycan. These results implicate ISPD in a-dystroglycan glycosylation in maintaining sarcolemma integrity in vertebrates.
Figures
References
-
- van Reeuwijk J, Brunner HG, van Bokhoven H. Glyc-O-genetics of Walker-Warburg syndrome. Clin Genet. 2005;67:281–9. - PubMed
-
- Kobayashi K, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388–92. - PubMed
-
- Yoshida A, et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. 2001;1:717–24. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
