

Systematic evaluation of factors influencing ChIP-seq fidelity
- PMID: 22522655
- PMCID: PMC3477507
- DOI: 10.1038/nmeth.1985
Systematic evaluation of factors influencing ChIP-seq fidelity
Abstract
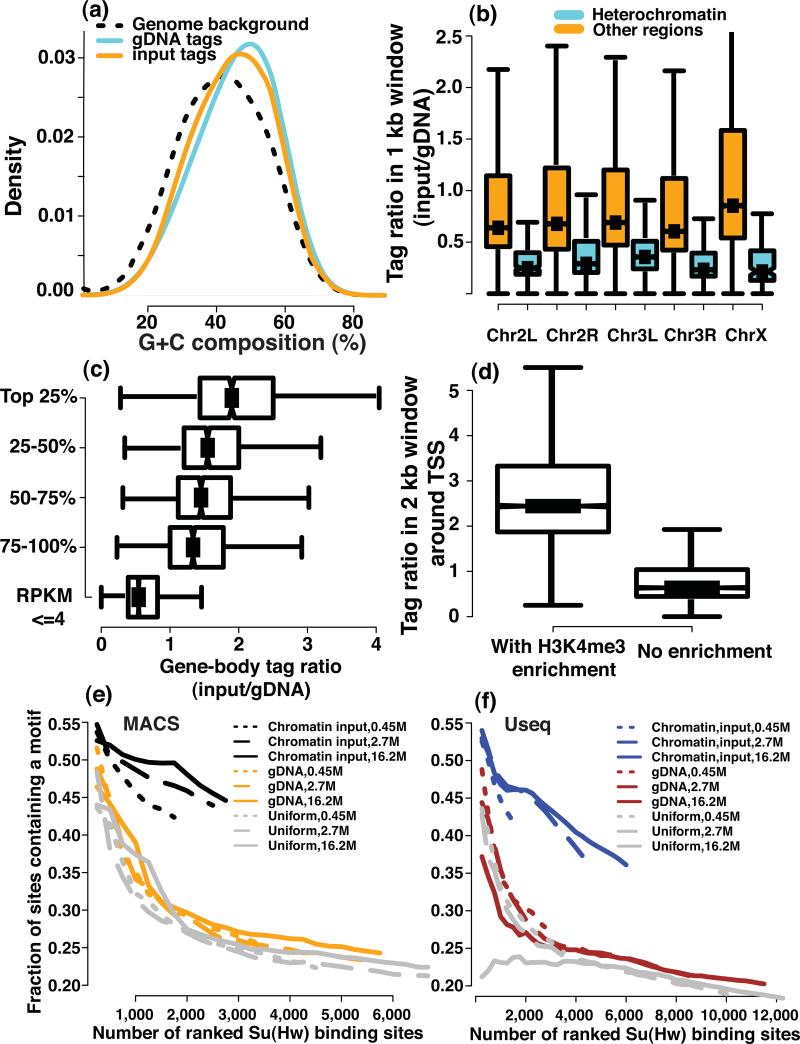
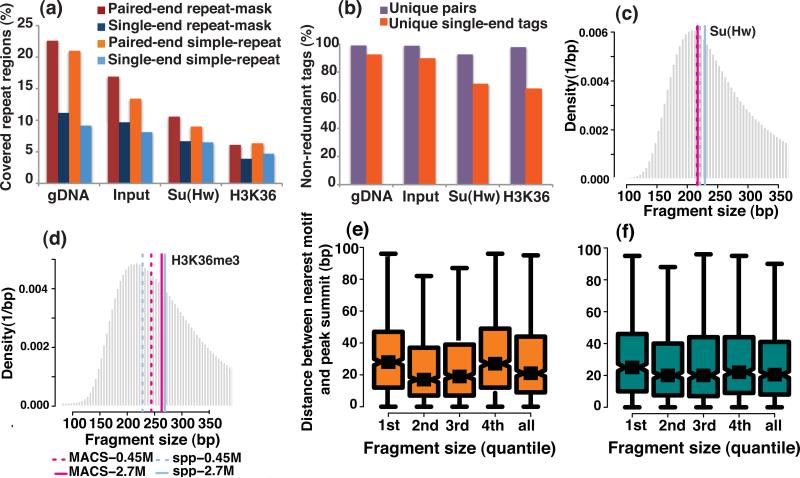
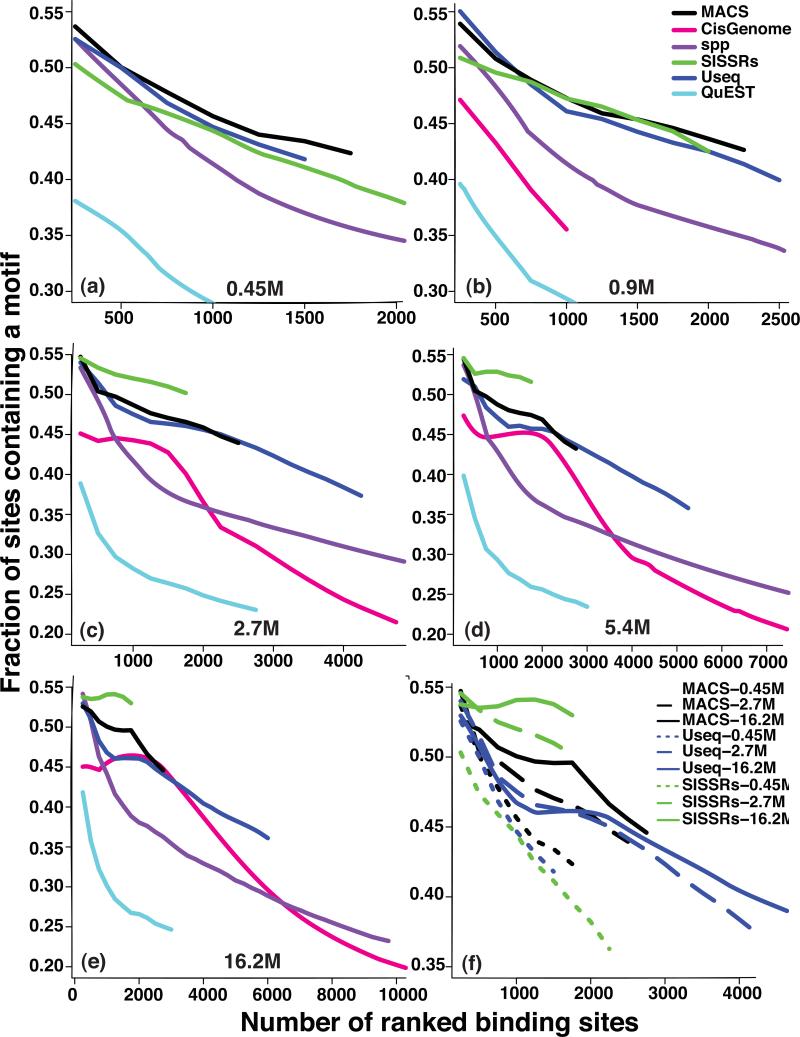
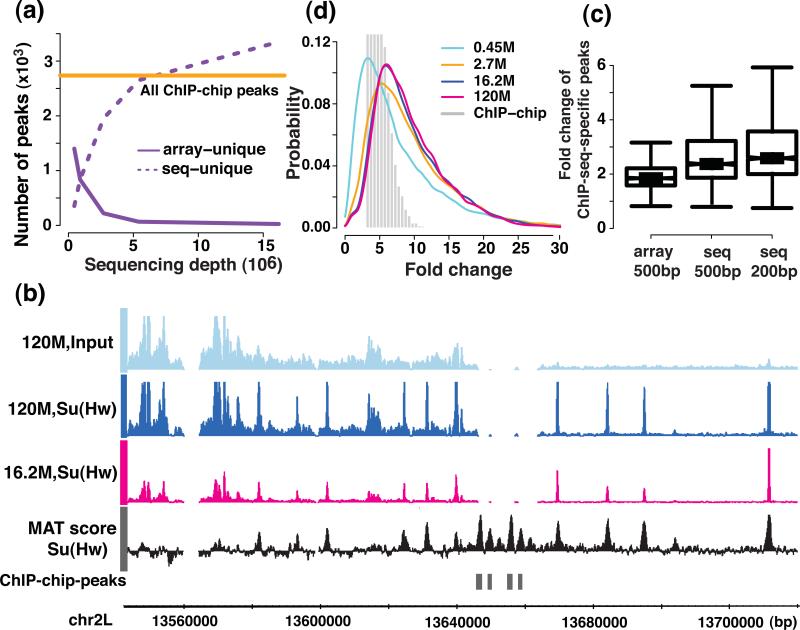
We evaluated how variations in sequencing depth and other parameters influence interpretation of chromatin immunoprecipitation-sequencing (ChIP-seq) experiments. Using Drosophila melanogaster S2 cells, we generated ChIP-seq data sets for a site-specific transcription factor (Suppressor of Hairy-wing) and a histone modification (H3K36me3). We detected a chromatin-state bias: open chromatin regions yielded higher coverage, which led to false positives if not corrected. This bias had a greater effect on detection specificity than any base-composition bias. Paired-end sequencing revealed that single-end data underestimated ChIP-library complexity at high coverage. Removal of reads originating at the same base reduced false-positives but had little effect on detection sensitivity. Even at mappable-genome coverage depth of ∼1 read per base pair, ∼1% of the narrow peaks detected on a tiling array were missed by ChIP-seq. Evaluation of widely used ChIP-seq analysis tools suggests that adjustments or algorithm improvements are required to handle data sets with deep coverage.
Figures

References
-
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. - PubMed
-
- Robertson G, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. - PubMed
-
- Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
