

Next generation sequencing for molecular diagnosis of neuromuscular diseases
- PMID: 22526018
- PMCID: PMC3400754
- DOI: 10.1007/s00401-012-0982-8
Next generation sequencing for molecular diagnosis of neuromuscular diseases
Abstract
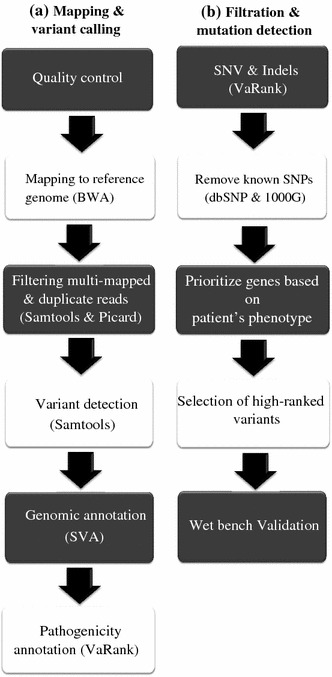
Inherited neuromuscular disorders (NMD) are chronic genetic diseases posing a significant burden on patients and the health care system. Despite tremendous research and clinical efforts, the molecular causes remain unknown for nearly half of the patients, due to genetic heterogeneity and conventional molecular diagnosis based on a gene-by-gene approach. We aimed to test next generation sequencing (NGS) as an efficient and cost-effective strategy to accelerate patient diagnosis. We designed a capture library to target the coding and splice site sequences of all known NMD genes and used NGS and DNA multiplexing to retrieve the pathogenic mutations in patients with heterogeneous NMD with or without known mutations. We retrieved all known mutations, including point mutations and small indels, intronic and exonic mutations, and a large deletion in a patient with Duchenne muscular dystrophy, validating the sensitivity and reproducibility of this strategy on a heterogeneous subset of NMD with different genetic inheritance. Most pathogenic mutations were ranked on top in our blind bioinformatic pipeline. Following the same strategy, we characterized probable TTN, RYR1 and COL6A3 mutations in several patients without previous molecular diagnosis. The cost was less than conventional testing for a single large gene. With appropriate adaptations, this strategy could be implemented into a routine genetic diagnosis set-up as a first screening approach to detect most kind of mutations, potentially before the need of more invasive and specific clinical investigations. An earlier genetic diagnosis should provide improved disease management and higher quality genetic counseling, and ease access to therapy or inclusion into therapeutic trials.
Figures


References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
