

Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population
- PMID: 22532803
- PMCID: PMC3330116
- DOI: 10.1371/journal.pgen.1002629
Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population
Abstract
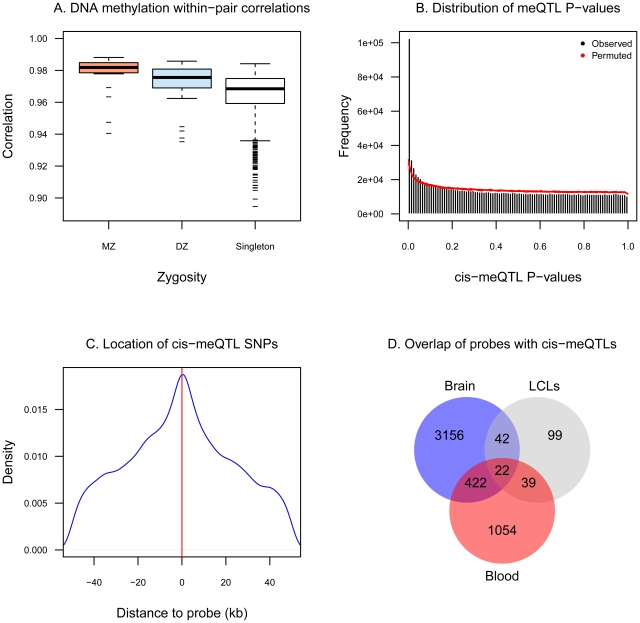
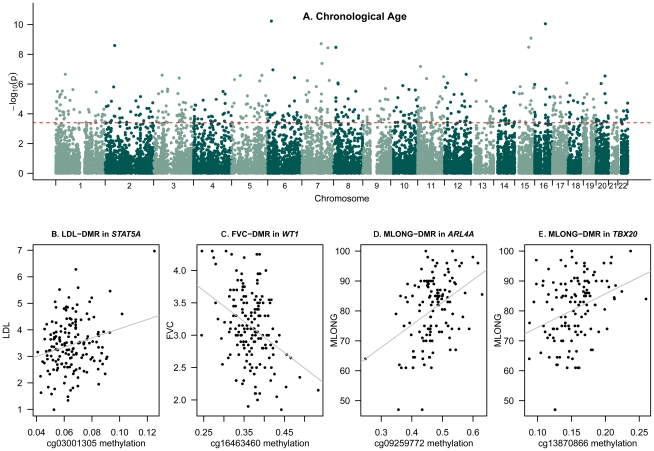
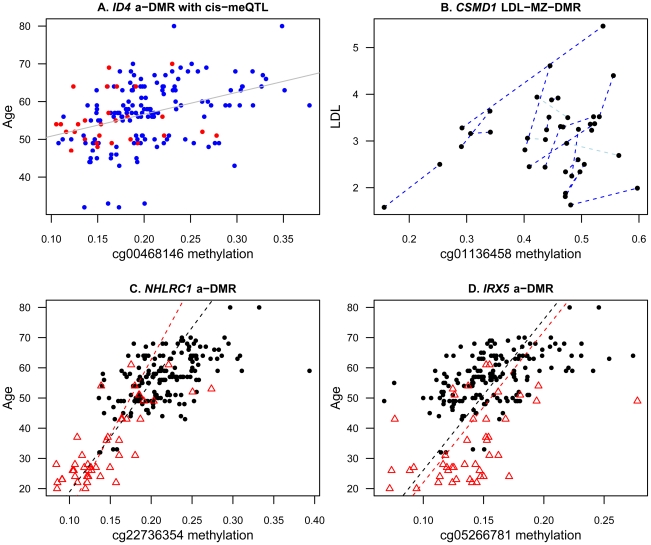
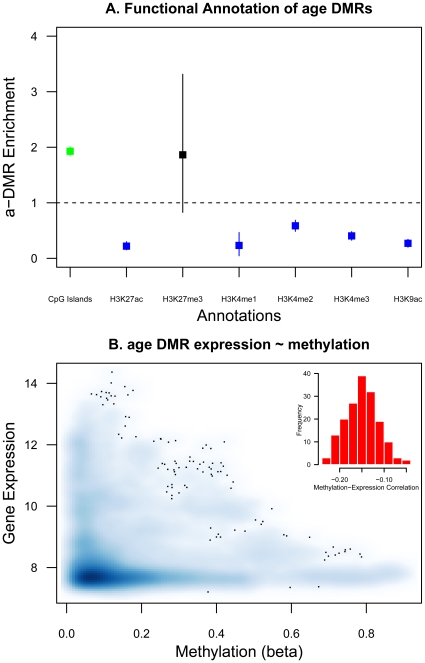
Age-related changes in DNA methylation have been implicated in cellular senescence and longevity, yet the causes and functional consequences of these variants remain unclear. To elucidate the role of age-related epigenetic changes in healthy ageing and potential longevity, we tested for association between whole-blood DNA methylation patterns in 172 female twins aged 32 to 80 with age and age-related phenotypes. Twin-based DNA methylation levels at 26,690 CpG-sites showed evidence for mean genome-wide heritability of 18%, which was supported by the identification of 1,537 CpG-sites with methylation QTLs in cis at FDR 5%. We performed genome-wide analyses to discover differentially methylated regions (DMRs) for sixteen age-related phenotypes (ap-DMRs) and chronological age (a-DMRs). Epigenome-wide association scans (EWAS) identified age-related phenotype DMRs (ap-DMRs) associated with LDL (STAT5A), lung function (WT1), and maternal longevity (ARL4A, TBX20). In contrast, EWAS for chronological age identified hundreds of predominantly hyper-methylated age DMRs (490 a-DMRs at FDR 5%), of which only one (TBX20) was also associated with an age-related phenotype. Therefore, the majority of age-related changes in DNA methylation are not associated with phenotypic measures of healthy ageing in later life. We replicated a large proportion of a-DMRs in a sample of 44 younger adult MZ twins aged 20 to 61, suggesting that a-DMRs may initiate at an earlier age. We next explored potential genetic and environmental mechanisms underlying a-DMRs and ap-DMRs. Genome-wide overlap across cis-meQTLs, genotype-phenotype associations, and EWAS ap-DMRs identified CpG-sites that had cis-meQTLs with evidence for genotype-phenotype association, where the CpG-site was also an ap-DMR for the same phenotype. Monozygotic twin methylation difference analyses identified one potential environmentally-mediated ap-DMR associated with total cholesterol and LDL (CSMD1). Our results suggest that in a small set of genes DNA methylation may be a candidate mechanism of mediating not only environmental, but also genetic effects on age-related phenotypes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. - DOI - PMC - PubMed
-
- Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41:240–245. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
