

Genome sequencing reveals complex speciation in the Drosophila simulans clade
- PMID: 22534282
- PMCID: PMC3409263
- DOI: 10.1101/gr.130922.111
Genome sequencing reveals complex speciation in the Drosophila simulans clade
Abstract
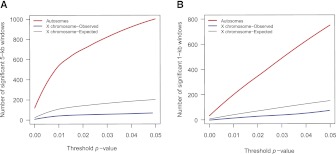
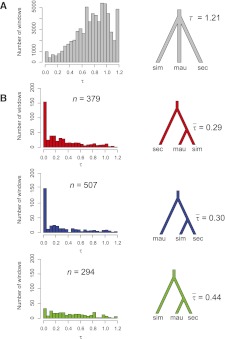
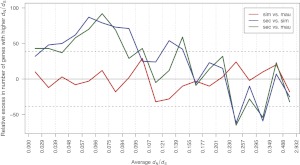
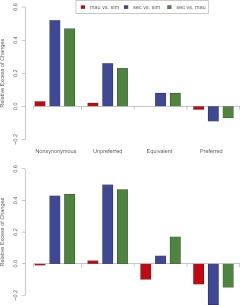
The three species of the Drosophila simulans clade--the cosmopolitan species, D. simulans, and the two island endemic species, D. mauritiana and D. sechellia--are important models in speciation genetics, but some details of their phylogenetic and speciation history remain unresolved. The order and timing of speciation are disputed, and the existence, magnitude, and timing of gene flow among the three species remain unclear. Here we report on the analysis of a whole-genome four-species sequence alignment that includes all three D. simulans clade species as well as the D. melanogaster reference sequence. The alignment comprises novel, paired short-read sequence data from a single highly inbred line each from D. simulans, D. mauritiana, and D. sechellia. We are unable to reject a species phylogeny with a basal polytomy; the estimated age of the polytomy is 242,000 yr before the present. However, we also find that up to 4.6% of autosomal and 2.2% of X-linked regions have evolutionary histories consistent with recent gene flow between the mainland species (D. simulans) and the two island endemic species (D. mauritiana and D. sechellia). Our findings thus show that gene flow has occurred throughout the genomes of the D. simulans clade species despite considerable geographic, ecological, and intrinsic reproductive isolation. Last, our analysis of lineage-specific changes confirms that the D. sechellia genome has experienced a significant excess of slightly deleterious changes and a dearth of presumed favorable changes. The relatively reduced efficacy of natural selection in D. sechellia is consistent with its derived, persistently reduced historical effective population size.
Figures


References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ 1990. Basic local alignment search tool. J Mol Biol 215: 403–410 - PubMed
-
- Aubert J, Solignac M 1990. Experimental evidence for mitochondrial DNA introgression between Drosophila species. Evolution 44: 1272–1282 - PubMed
-
- Ballard JW 2000a. Comparative genomics of mitochondrial DNA in members of the Drosophila melanogaster subgroup. J Mol Evol 51: 48–63 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases