

Matricellular proteins in cardiac adaptation and disease
- PMID: 22535894
- PMCID: PMC4411042
- DOI: 10.1152/physrev.00008.2011
Matricellular proteins in cardiac adaptation and disease
Abstract
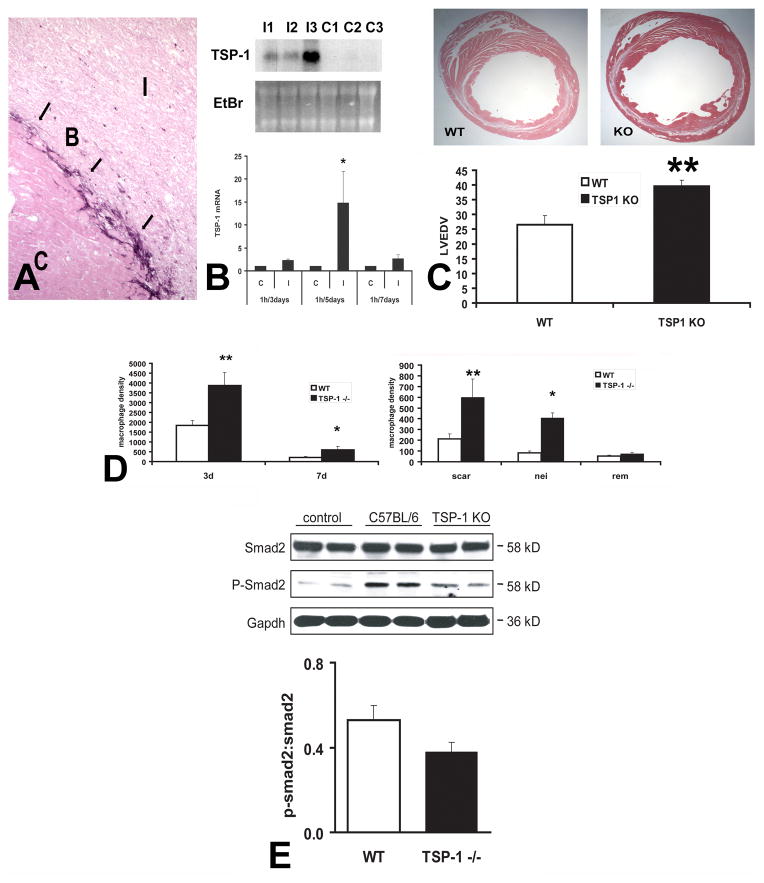
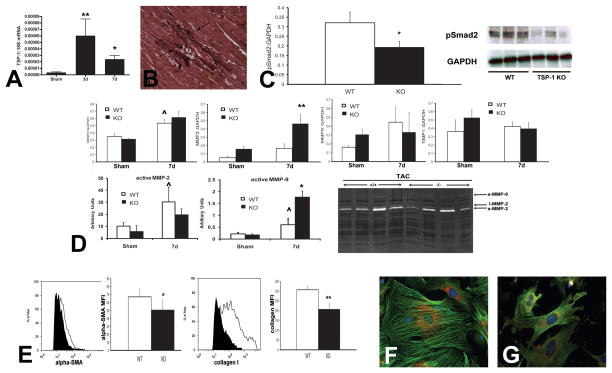
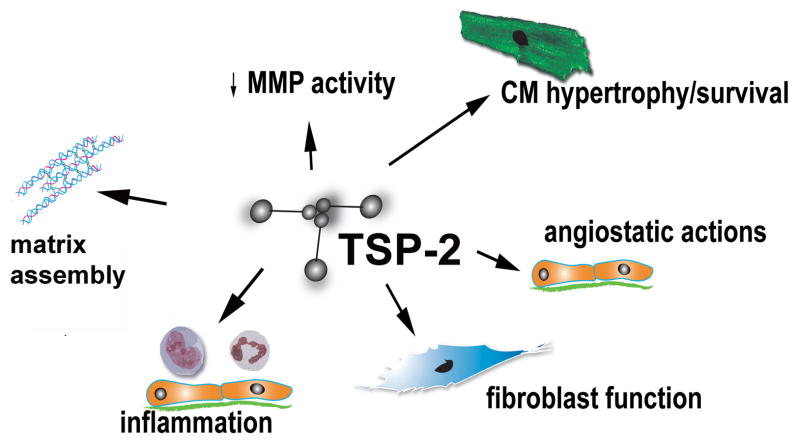
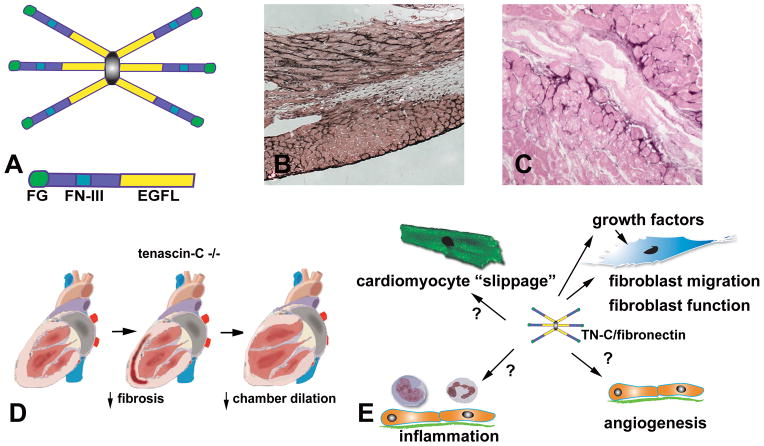
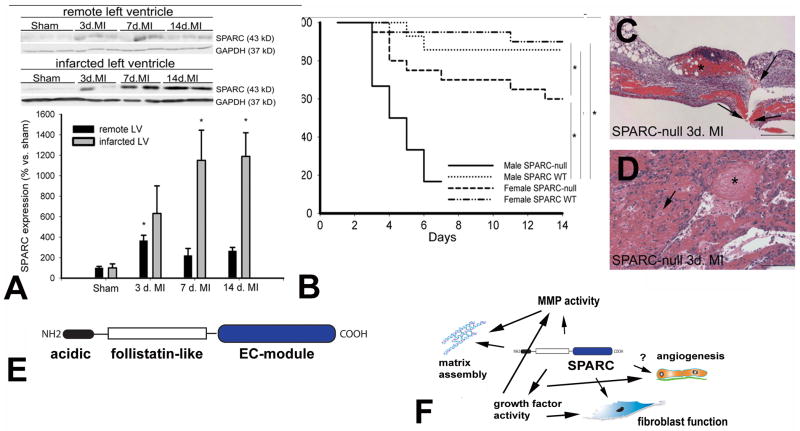
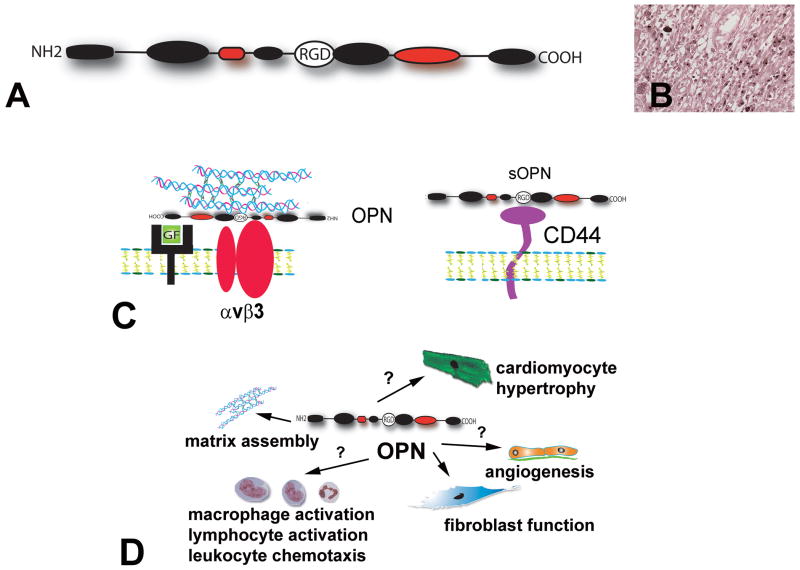
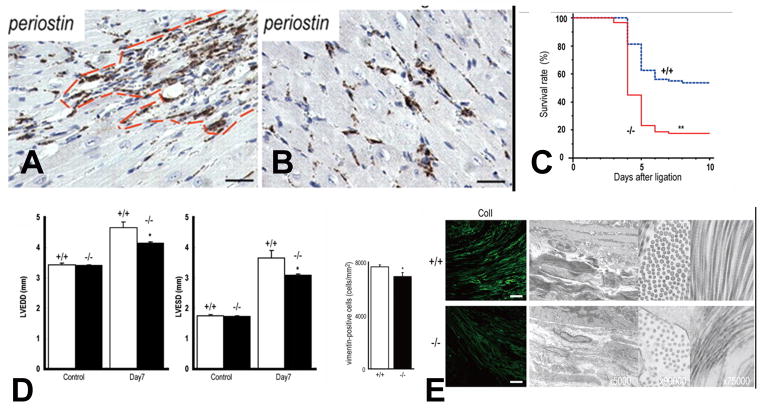
The term matricellular proteins describes a family of structurally unrelated extracellular macromolecules that, unlike structural matrix proteins, do not play a primary role in tissue architecture, but are induced following injury and modulate cell-cell and cell-matrix interactions. When released to the matrix, matricellular proteins associate with growth factors, cytokines, and other bioactive effectors and bind to cell surface receptors transducing signaling cascades. Matricellular proteins are upregulated in the injured and remodeling heart and play an important role in regulation of inflammatory, reparative, fibrotic and angiogenic pathways. Thrombospondin (TSP)-1, -2, and -4 as well as tenascin-C and -X secreted protein acidic and rich in cysteine (SPARC), osteopontin, periostin, and members of the CCN family (including CCN1 and CCN2/connective tissue growth factor) are involved in a variety of cardiac pathophysiological conditions, including myocardial infarction, cardiac hypertrophy and fibrosis, aging-associated myocardial remodeling, myocarditis, diabetic cardiomyopathy, and valvular disease. This review discusses the properties and characteristics of the matricellular proteins and presents our current knowledge on their role in cardiac adaptation and disease. Understanding the role of matricellular proteins in myocardial pathophysiology and identification of the functional domains responsible for their actions may lead to design of peptides with therapeutic potential for patients with heart disease.
Figures
References
-
- Abe K, Hibino T, Mishima H, Shimomura Y. The cytokine regulation of SPARC production by rabbit corneal epithelial cells and fibroblasts in vitro. Cornea. 2004;23:172–179. - PubMed
-
- Abraham D. Connective tissue growth factor: growth factor, matricellular organizer, fibrotic biomarker or molecular target for anti-fibrotic therapy in SSc? Rheumatology (Oxford) 2008;47 (Suppl 5):v8–9. - PubMed
-
- Adams JC. Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol. 2001;17:25–51. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous