

KLF15 negatively regulates estrogen-induced epithelial cell proliferation by inhibition of DNA replication licensing
- PMID: 22538816
- PMCID: PMC3361390
- DOI: 10.1073/pnas.1118515109
KLF15 negatively regulates estrogen-induced epithelial cell proliferation by inhibition of DNA replication licensing
Abstract
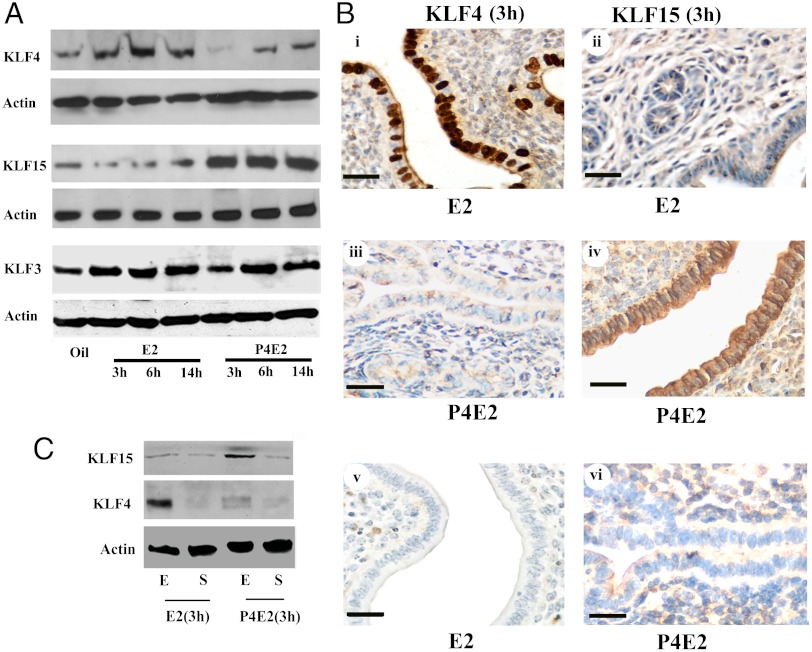
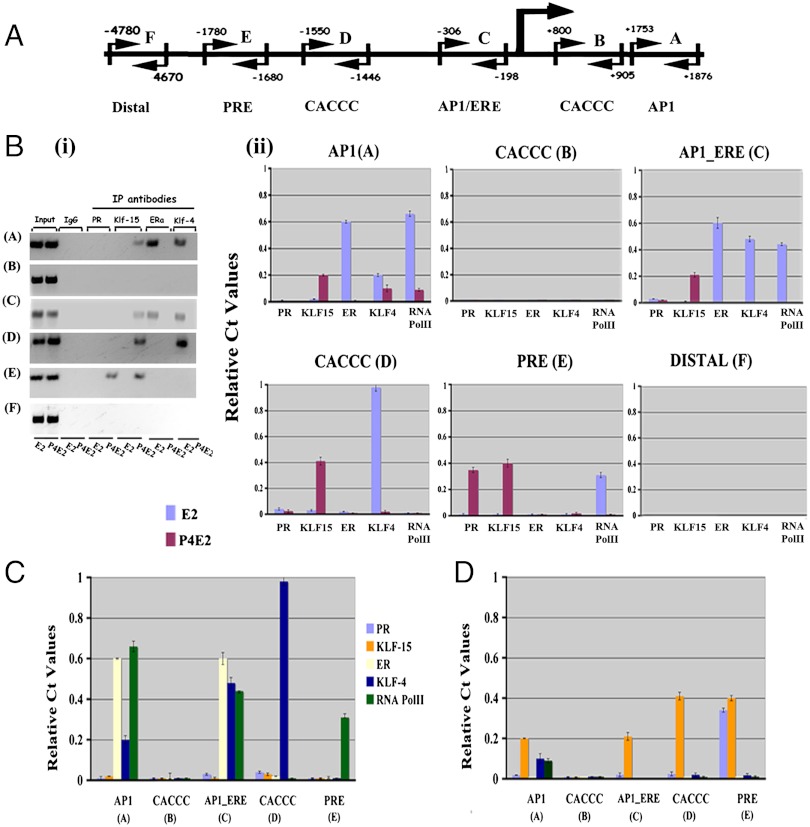
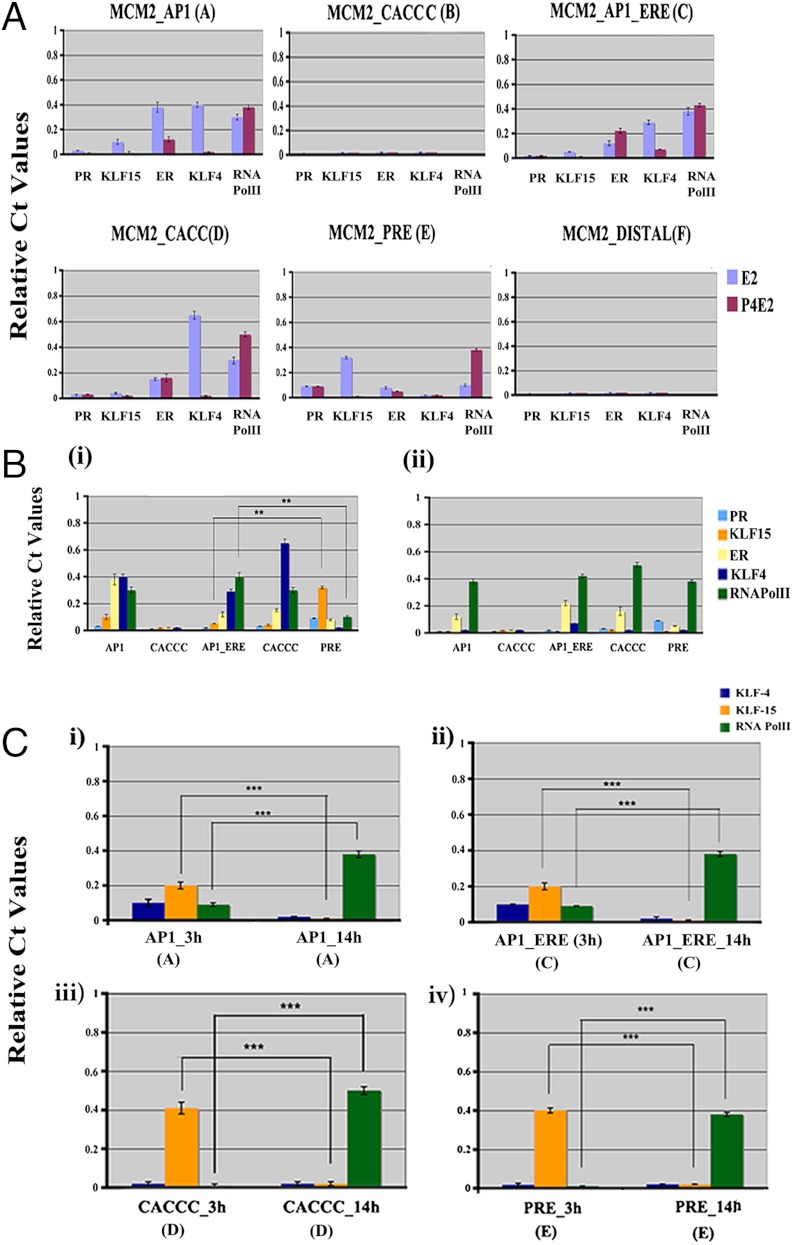
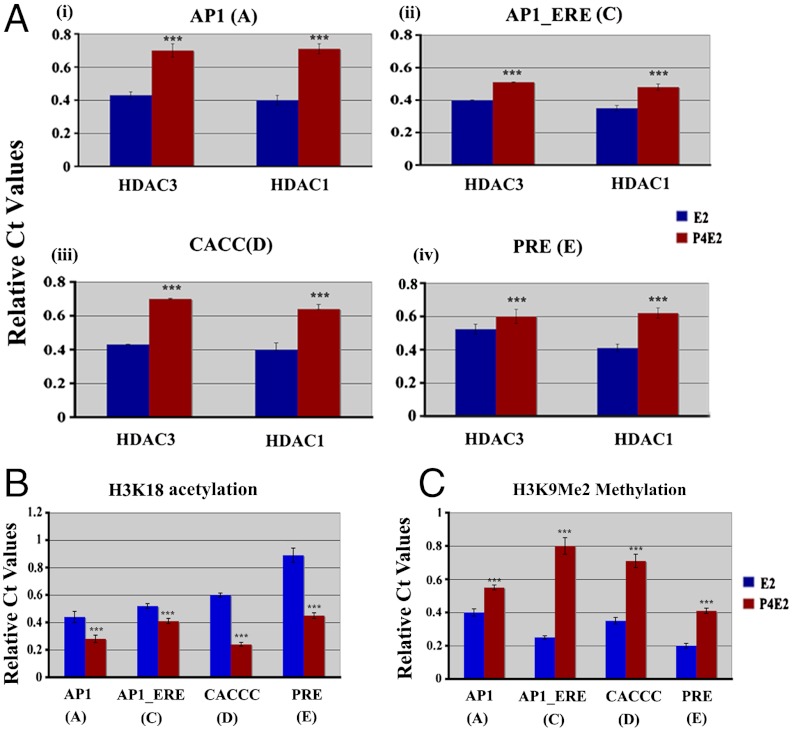
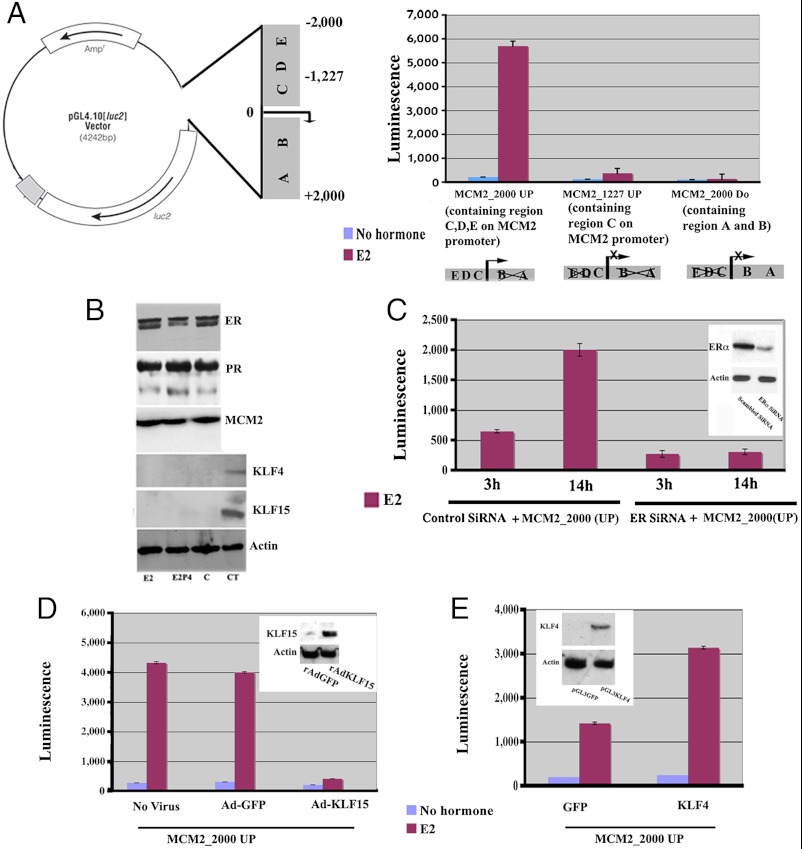
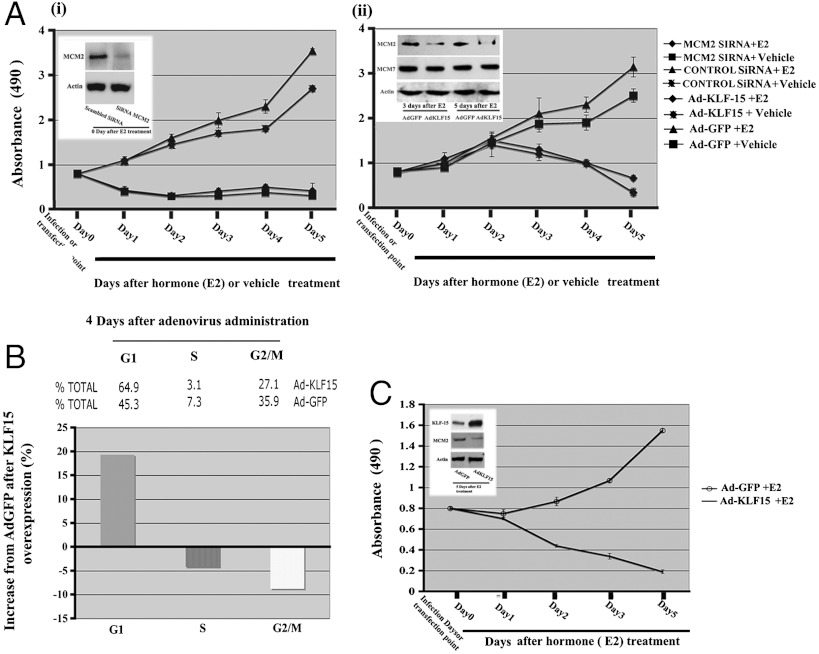
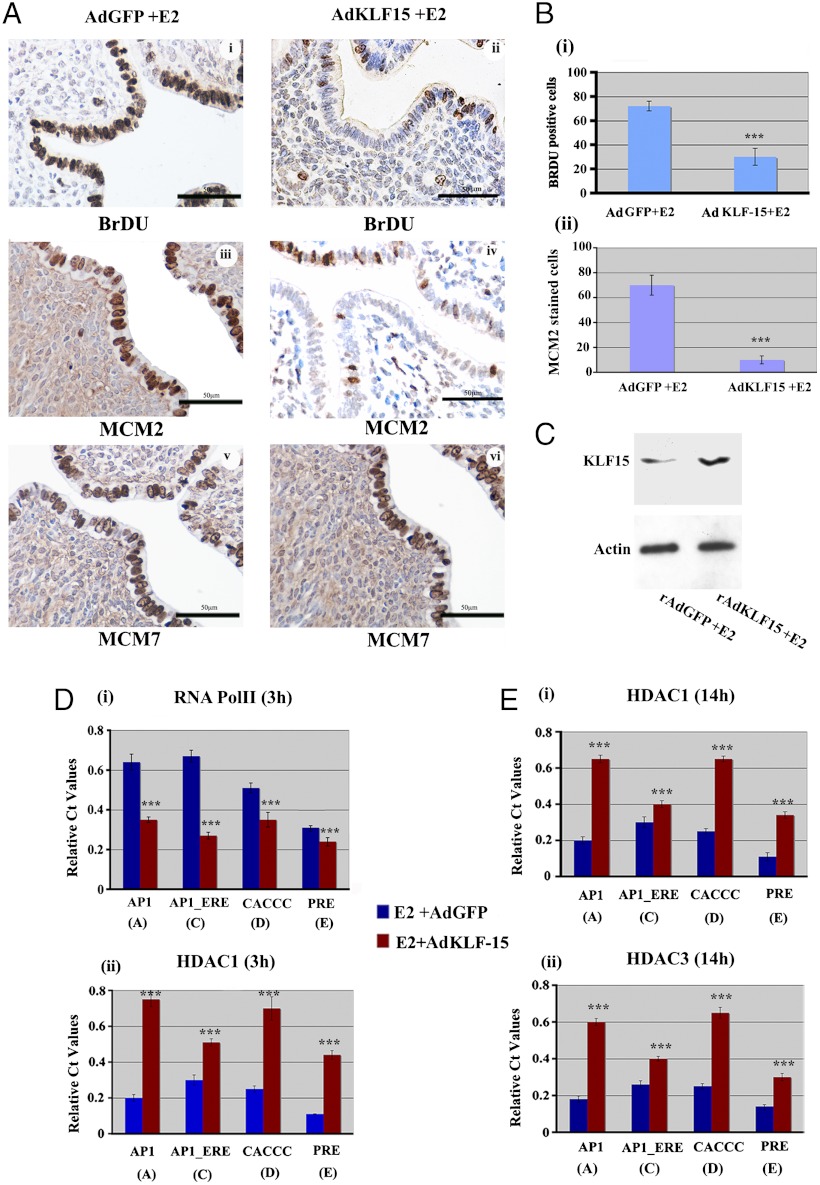
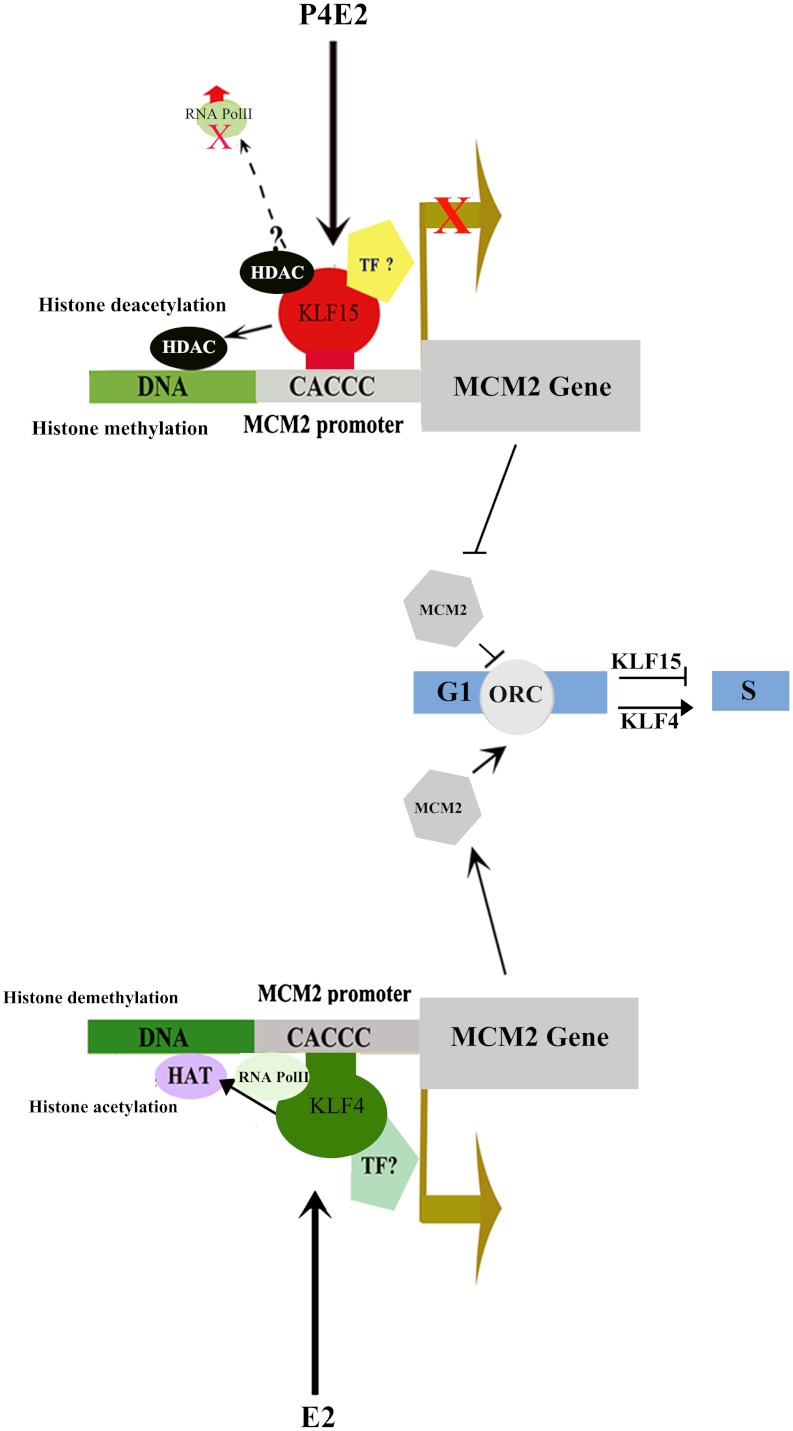
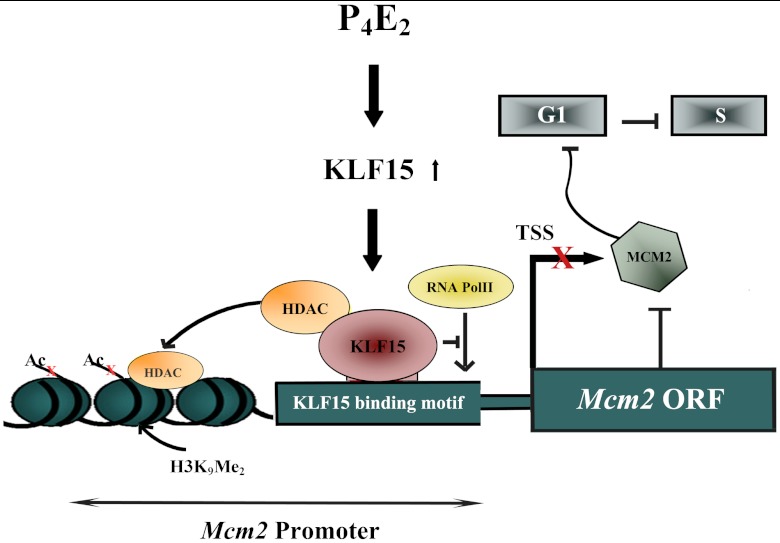
In the epithelial compartment of the uterus, estradiol-17β (E(2)) induces cell proliferation while progesterone (P(4)) inhibits this response and causes differentiation of the cells. In this study, we identified the mechanism whereby E(2) and P(4) reciprocally regulate the expression of minichromosome maintenance (MCM)-2, a protein that is an essential component of the hexameric MCM-2 to 7 complex required for DNA synthesis initiation. We show in the uterine epithelium that Kruppel-like transcription (KLF) factors, KLF 4 and 15, are inversely expressed; most importantly, they bind to the Mcm2 promoter under the regulation of E(2) and P(4)E(2), respectively. After P(4)E(2) exposure and in contrast to E(2) treated mice, the Mcm2 promoter displays increased histone 3 (H3) methylation and the recruitment of histone deacetylase 1 and 3 with the concomitant deacetylation of H3. This increased methylation and decreased acetylation is associated with an inhibition of RNA polymerase II binding, indicating an inactive Mcm2 promoter following P(4)E(2) treatment. Using transient transfection assays in the Ishikawa endometrial cell line, we demonstrate that Mcm2 promoter activity is hormonally stimulated by E(2) and that KLF15 inhibits this E(2) enhanced transcription. KLF15 expression also blocks Ishikawa cell proliferation through inhibition of MCM2 protein level. Importantly, in vivo expression of KLF15 in an estrogenized uterus mimics P(4)'s action by inhibiting E(2)-induced uterine epithelial MCM-2 expression and DNA synthesis. KLF15 is therefore a downstream physiological mediator of progesterone's cell cycle inhibitory action in the uterine epithelium.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Rubel CA, Jeong JW, Tsai SY, Lydon JP, Demayo FJ. Epithelial-stromal interaction and progesterone receptors in the mouse uterus. Semin Reprod Med. 2010;28:27–35. - PubMed
-
- Korhonen MO, Symons JP, Hyde BM, Rowan JP, Wilborn WH. Histologic classification and pathologic findings for endometrial biopsy specimens obtained from 2,964 perimenopausal and postmenopausal women undergoing screening for continuous hormones as replacement therapy (CHART 2 Study) Am J Obstet Gynecol. 1997;176:377–380. - PubMed
-
- Pike MC, et al. Estrogen-progestin replacement therapy and endometrial cancer. J Natl Cancer Inst. 1997;89:1110–1116. - PubMed
-
- Tong W, et al. Female sex steroid hormone regulation of cell proliferation in the endometrium. In: Aplin JDFS, Glasser SR, Gludice LC, editors. The Endometrium: Molecular, Cellular and Clinical Persectives. 2nd Ed. London: Taylor and Francis; 2008. pp. 99–122.
-
- Niklaus AL, et al. Assessment of the proliferative status of epithelial cell types in the endometrium of young and menopausal transition women. Hum Reprod. 2007;22:1778–1788. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
