

Increased activation of hereditary pancreatitis-associated human cationic trypsinogen mutants in presence of chymotrypsin C
- PMID: 22539344
- PMCID: PMC3370252
- DOI: 10.1074/jbc.M112.360065
Increased activation of hereditary pancreatitis-associated human cationic trypsinogen mutants in presence of chymotrypsin C
Abstract
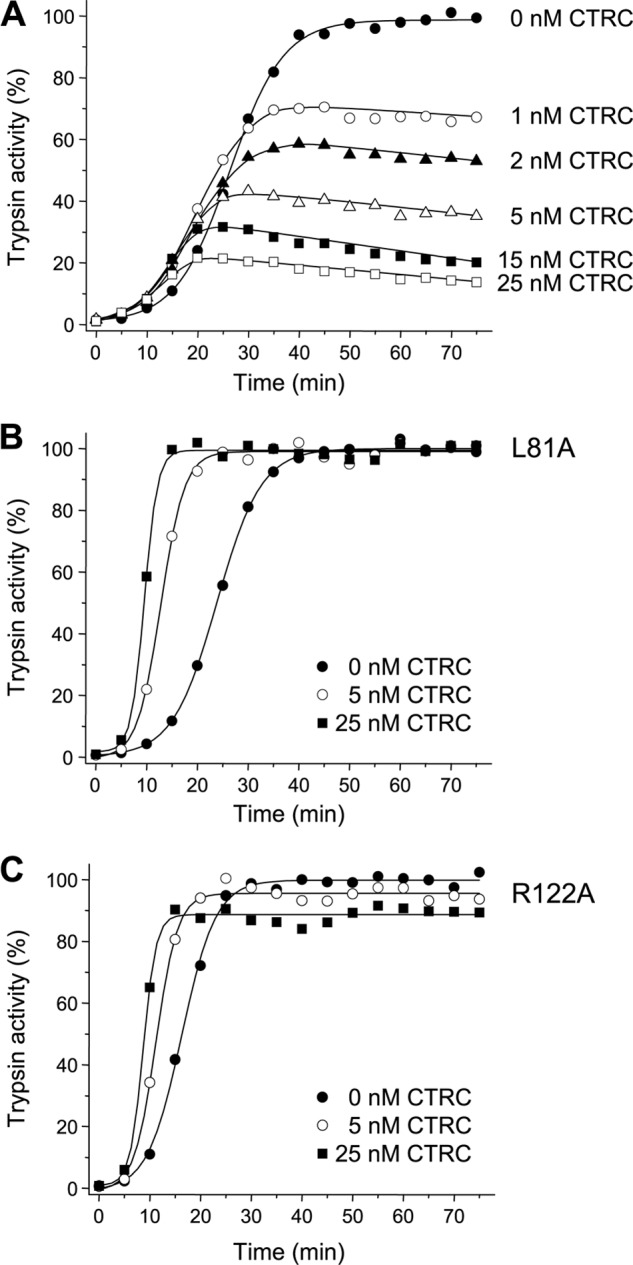
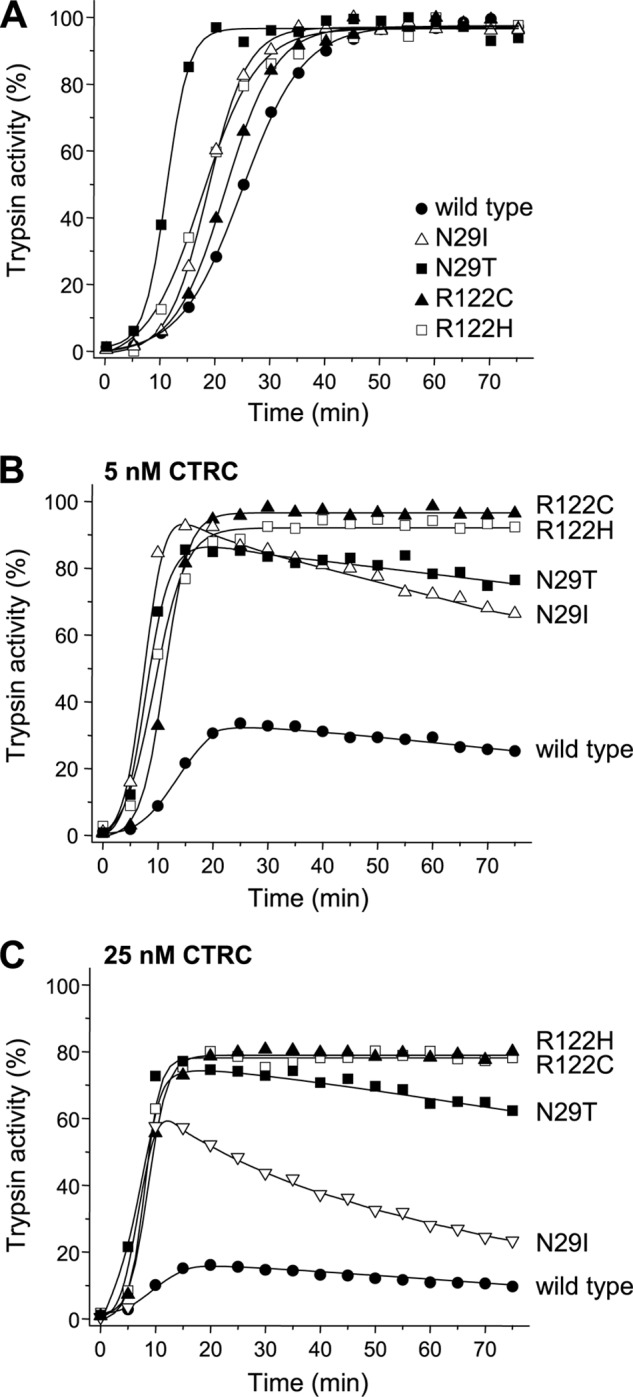
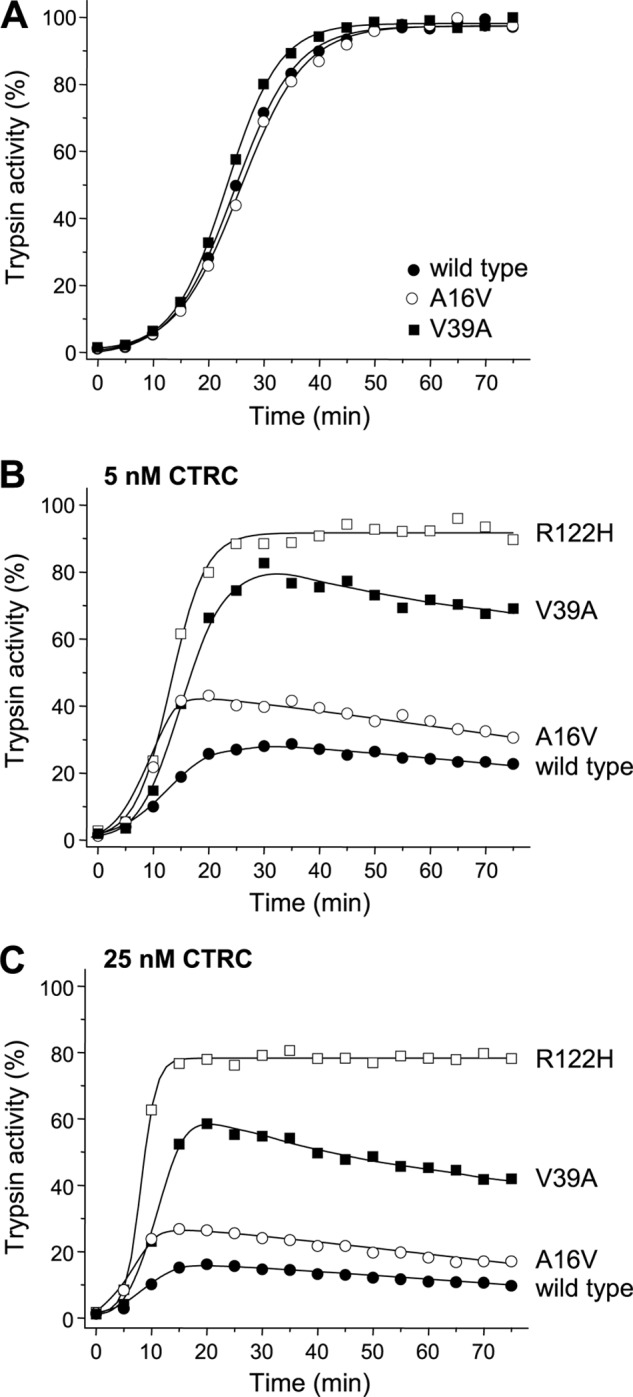
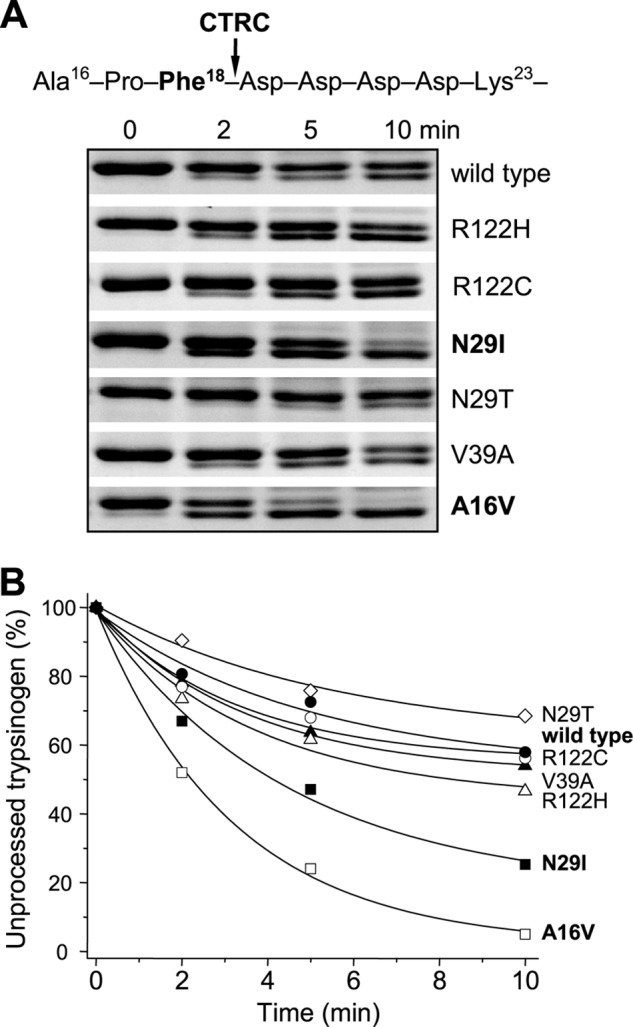
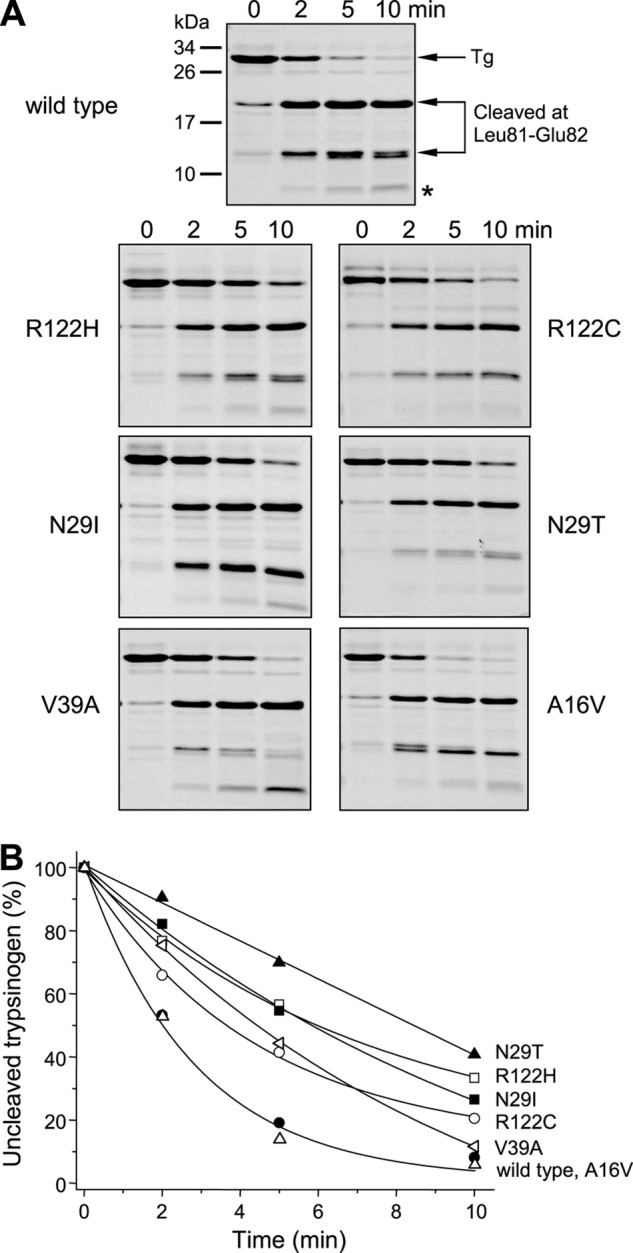
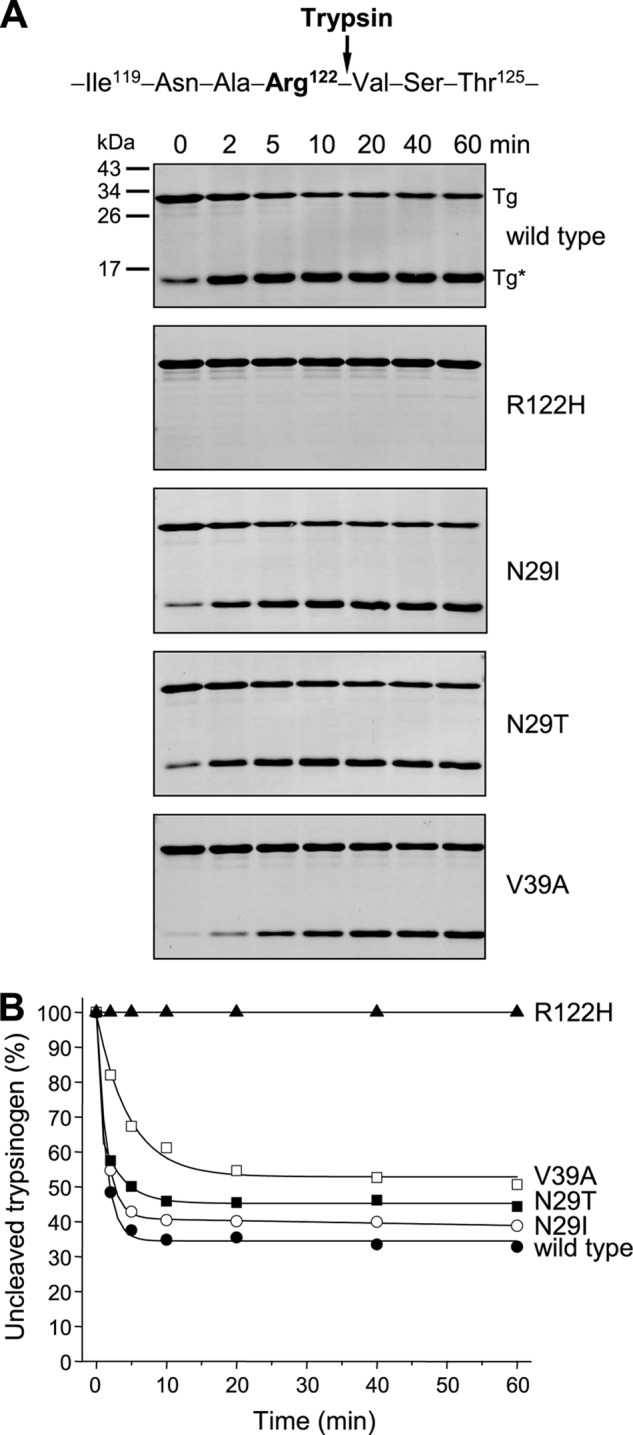
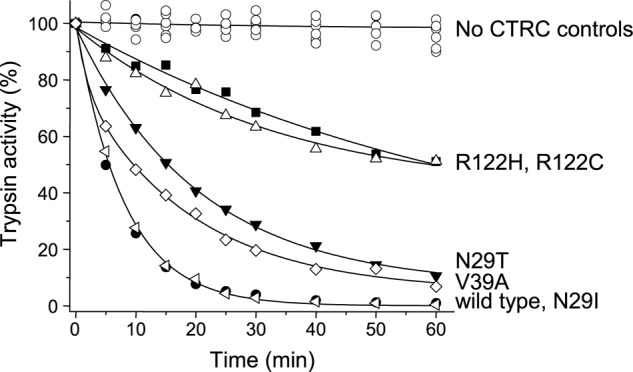
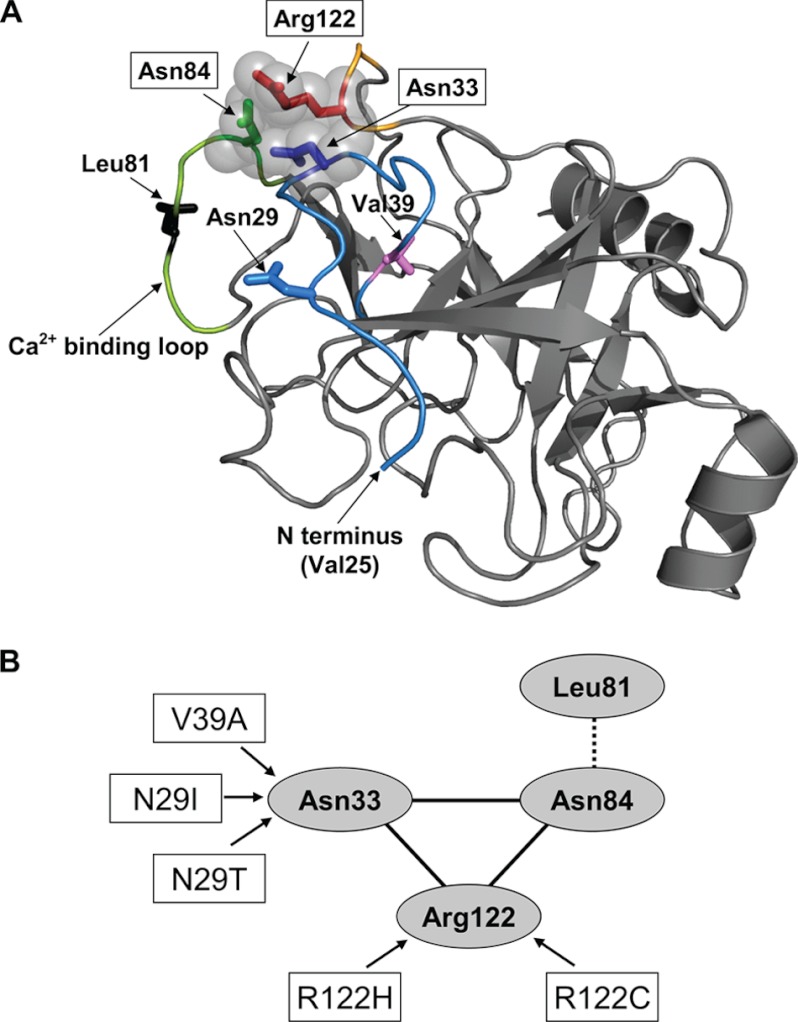
Mutations in human cationic trypsinogen (PRSS1) cause autosomal dominant hereditary pancreatitis. Increased intrapancreatic autoactivation of trypsinogen mutants has been hypothesized to initiate the disease. Autoactivation of cationic trypsinogen is proteolytically regulated by chymotrypsin C (CTRC), which mitigates the development of trypsin activity by promoting degradation of both trypsinogen and trypsin. Paradoxically, CTRC also increases the rate of autoactivation by processing the trypsinogen activation peptide to a shorter form. The aim of this study was to investigate the effect of CTRC on the autoactivation of clinically relevant trypsinogen mutants. We found that in the presence of CTRC, trypsinogen mutants associated with classic hereditary pancreatitis (N29I, N29T, V39A, R122C, and R122H) autoactivated at increased rates and reached markedly higher active trypsin levels compared with wild-type cationic trypsinogen. The A16V mutant, known for its variable disease penetrance, exhibited a smaller increase in autoactivation. The mechanistic basis of increased activation was mutation-specific and involved resistance to degradation (N29I, N29T, V39A, R122C, and R122H) and/or increased N-terminal processing by CTRC (A16V and N29I). These observations indicate that hereditary pancreatitis is caused by CTRC-dependent dysregulation of cationic trypsinogen autoactivation, which results in elevated trypsin levels in the pancreas.
Figures
References
-
- Whitcomb D. C., Gorry M. C., Preston R. A., Furey W., Sossenheimer M. J., Ulrich C. D., Martin S. P., Gates L. K., Jr., Amann S. T., Toskes P. P., Liddle R., McGrath K., Uomo G., Post J. C., Ehrlich G. D. (1996) Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet. 14, 141–145 - PubMed
-
- Howes N., Lerch M. M., Greenhalf W., Stocken D. D., Ellis I., Simon P., Truninger K., Ammann R., Cavallini G., Charnley R. M., Uomo G., Delhaye M., Spicak J., Drumm B., Jansen J., Mountford R., Whitcomb D. C., Neoptolemos J. P., and European Registry of Hereditary Pancreatitis and Pancreatic Cancer (EUROPAC) (2004) Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin. Gastroenterol. Hepatol. 2, 252–261 - PubMed
-
- Rebours V., Boutron-Ruault M. C., Schnee M., Férec C., Le Maréchal C., Hentic O., Maire F., Hammel P., Ruszniewski P., Lévy P. (2009) The natural history of hereditary pancreatitis: a national series. Gut 58, 97–103 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
