

The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post-Golgi trafficking
- PMID: 22545070
- PMCID: PMC3334816
- DOI: 10.2217/clp.11.70
The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post-Golgi trafficking
Abstract
Loss-of-function mutations in CLN3 are responsible for juvenile-onset neuronal ceroid lipofuscinosis (JNCL), or Batten disease, which is an incurable lysosomal disease that manifests with vision loss, followed by seizures and progressive neurodegeneration, robbing children of motor skills, speech and cognition, and eventually leading to death in the second or third decade of life. Emerging clinical evidence points to JNCL pathology outside of the CNS, including the cardiovascular system. The CLN3 gene encodes an unusual transmembrane protein, CLN3 or battenin, whose elusive function has been the subject of intense study for more than 10 years. Owing to the detailed characterization of a large number of disease models, our knowledge of CLN3 protein function is finally coming into focus. This review will describe the most current understanding of CLN3 structure, function and dysfunction in JNCL.
Figures
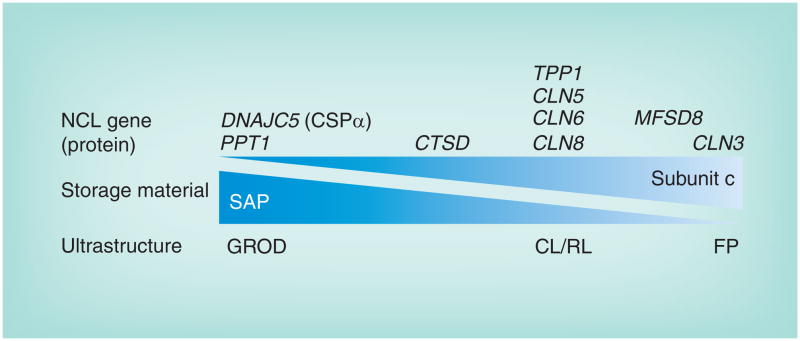


References
-
- Kohlschutter A, Schulz A. Towards understanding the neuronal ceroid lipofuscinoses. Brain Dev. 2009;31(7):499–502. - PubMed
-
- Jalanko A, Braulke T. Neuronal ceroid lipofuscinoses. Biochim Biophys Acta. 2009;1793(4):697–709. - PubMed
-
- Mole S, Williams R, Goebel H. The Neuronal Ceroid Lipofuscinoses (Batten Disease) 2. Oxford University Press; UK: 2011. Comprehensive review of the clinicopathologic features of neuronal ceroid lipofuscinosis (NCL), the spectrum of mutations in each of the NCL genes known at the time of its publication, the genetic models that have been developed for each form of the disease and the proposed functions of each of the proteins encoded by the NCL genes.
-
- Mole S, Williams R, Goebel H. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6(3):107–126. - PubMed
-
- Chattopadhyay S, Ito M, Cooper J, et al. An autoantibody inhibitory to glutamic acid decarboxylase in the neurodegenerative disorder Batten disease. Hum Mol Genet. 2002;11(12):1421–1431. - PubMed
Websites
-
- SCOP: Structural Classification of Proteins, 1.75 release. 2009 Jun; http://scop.mrc-lmb.cam.ac.uk/scop.
-
- The Wellcome Trust. Sanger Institute. Pfam 26.0. 2011 Nov; http://pfam.sanger.ac.uk.
-
- NCL Resource – A gateway for Batten disease. www.ucl.ac.uk/ncl.
-
- UCSC Genome Bioinformatics. http://genome.ucsc.edu.
-
- Gene expression omnibus. www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE24368.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical