

Metal-catalyzed nitrogen-atom transfer methods for the oxidation of aliphatic C-H bonds
- PMID: 22546004
- PMCID: PMC5483381
- DOI: 10.1021/ar200318q
Metal-catalyzed nitrogen-atom transfer methods for the oxidation of aliphatic C-H bonds
Abstract
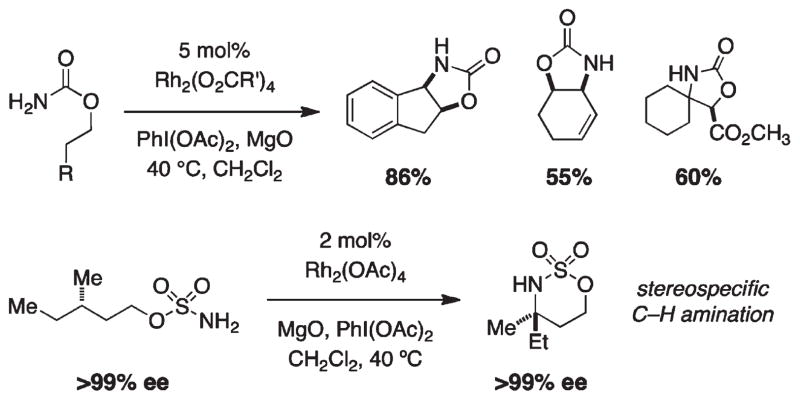
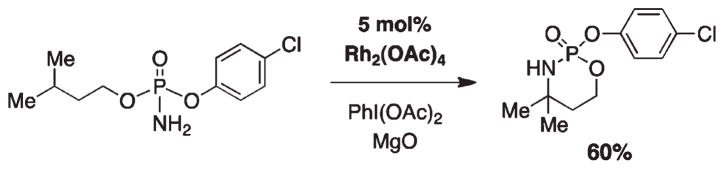
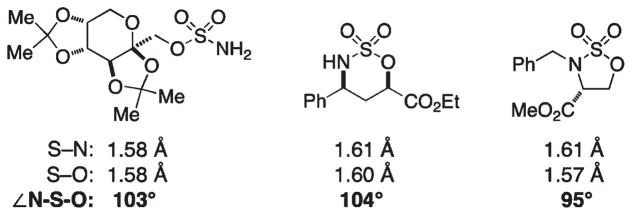
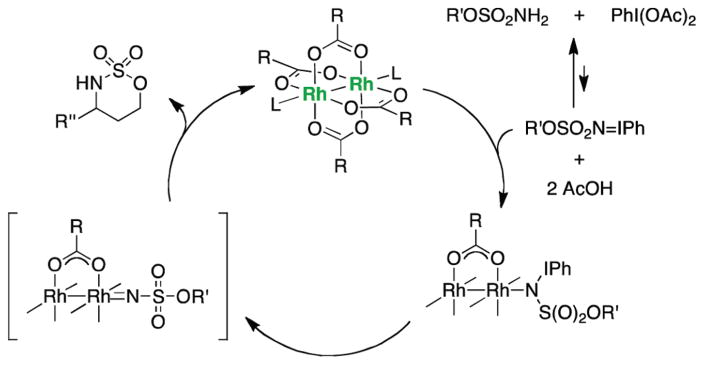
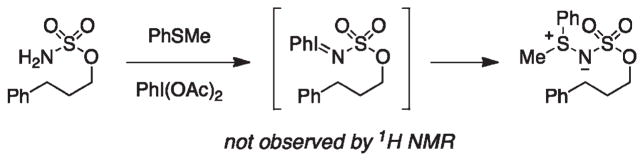
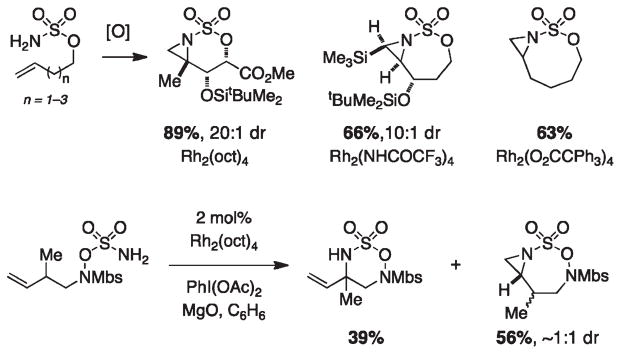
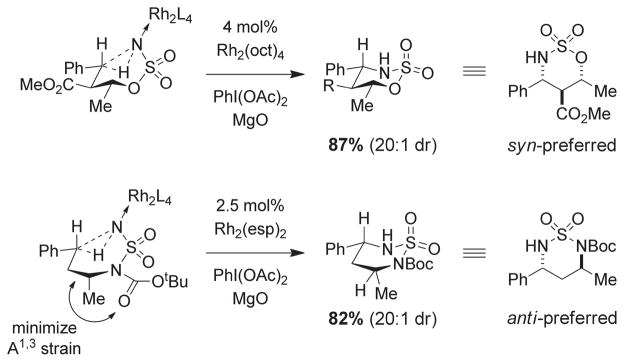
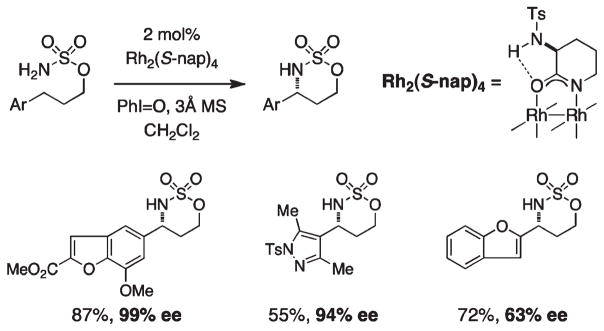
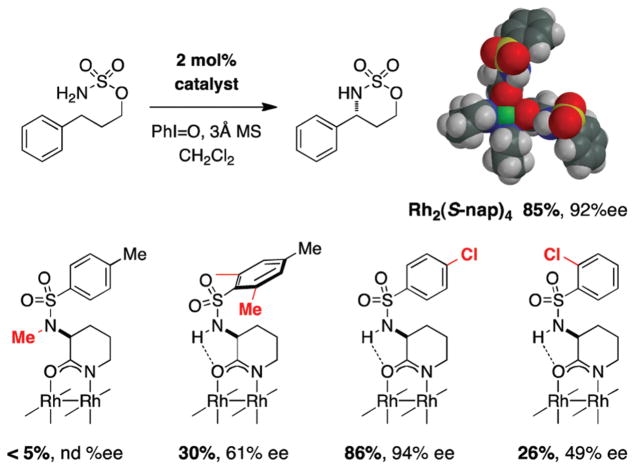
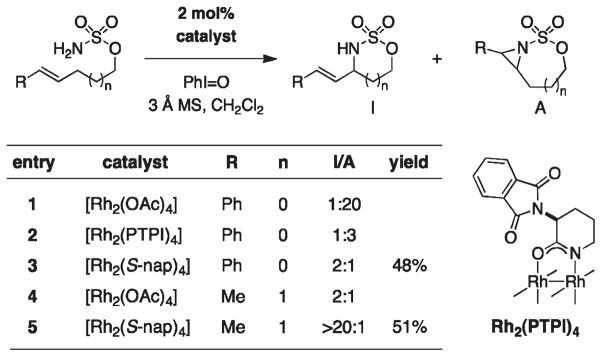
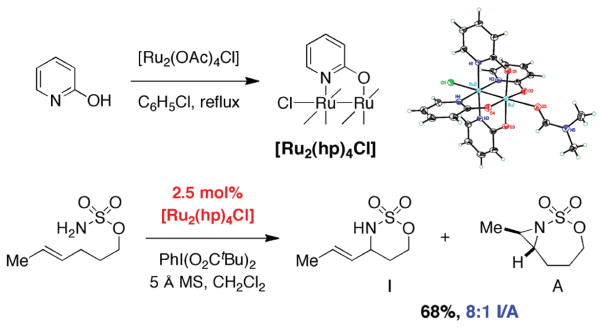
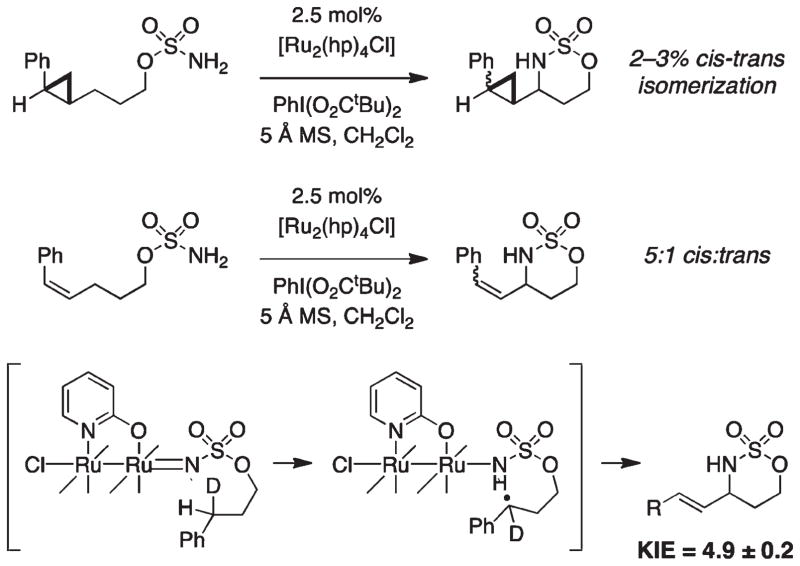
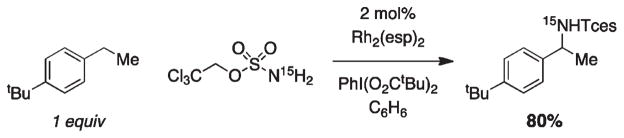
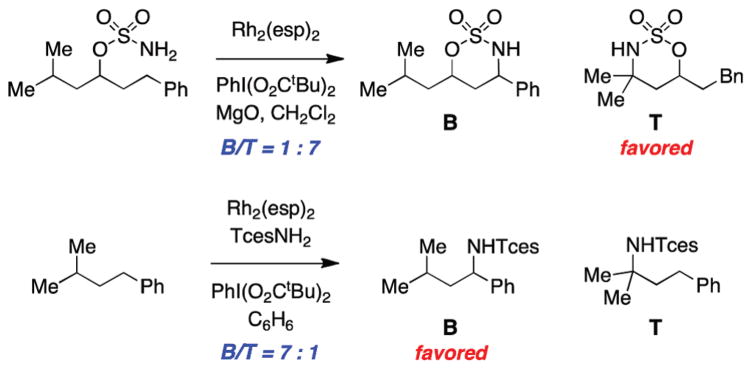
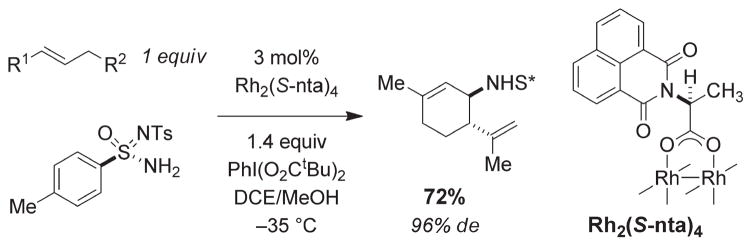
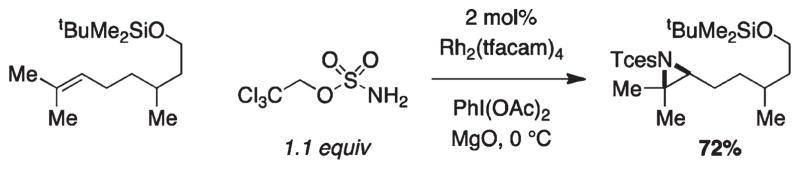
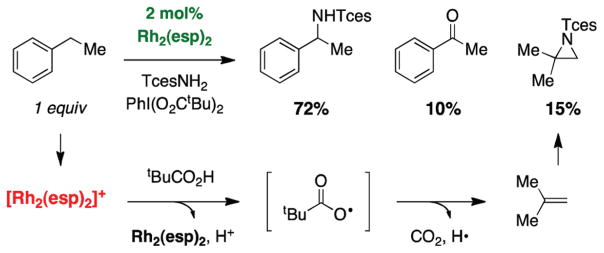
For more than a century, chemists have endeavored to discover and develop reaction processes that enable the selective oxidation of hydrocarbons. In the 1970s, Abramovitch and Yamada described the synthesis and electrophilic reactivity of sulfonyliminoiodinanes (RSO(2)N═IPh), demonstrating the utility of this new class of reagents to function as nitrene equivalents. Subsequent investigations by Breslow, Mansuy, and Müller would show such oxidants to be competent for alkene and saturated hydrocarbon functionalization when combined with transition metal salts or metal complexes, namely those of Mn, Fe, and Rh. Here, we trace our own studies to develop N-atom transfer technologies for C-H and π-bond oxidation. This Account discusses advances in both intra- and intermolecular amination processes mediated by dirhodium and diruthenium complexes, as well as the mechanistic foundations of catalyst reactivity and arrest. Explicit reference is given to questions that remain unanswered and to problem areas that are rich for discovery. A fundamental advance in amination technology has been the recognition that iminoiodinane oxidants can be generated in situ in the presence of a metal catalyst that elicits subsequent N-atom transfer. Under these conditions, both dirhodium and diruthenium lantern complexes function as competent catalysts for C-H bond oxidation with a range of nitrogen sources (e.g., carbamates, sulfamates, sulfamides, etc.), many of which will not form isolable iminoiodinane equivalents. Practical synthetic methods and applications thereof have evolved in parallel with inquiries into the operative reaction mechanism(s). For the intramolecular dirhodium-catalyzed process, the body of experimental and computational data is consistent with a concerted asynchronous C-H insertion pathway, analogous to the consensus mechanism for Rh-carbene transfer. Other studies reveal that the bridging tetracarboxylate ligand groups, which shroud the dirhodium core, are labile to exchange under standard reaction conditions. This information has led to the generation of chelating dicarboxylate dinuclear rhodium complexes, exemplified by Rh(2)(esp)(2). The performance of this catalyst system is unmatched by other dirhodium complexes in both intra- and intermolecular C-H amination reactions. Tetra-bridged, mixed-valent diruthenium complexes function as effective promoters of sulfamate ester oxidative cyclization. These catalysts can be crafted with ligand sets other than carboxylates and are more resistant to oxidation than their dirhodium counterparts. A range of experimental and computational mechanistic data amassed with the tetra-2-oxypyridinate diruthenium chloride complex, [Ru(2)(hp)(4)Cl], has established the insertion event as a stepwise pathway involving a discrete radical intermediate. These data contrast dirhodium-catalyzed C-H amination and offer a cogent model for understanding the divergent chemoselectivity trends observed between the two catalyst types. This work constitutes an important step toward the ultimate goal of achieving predictable, reagent-level control over product selectivity.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
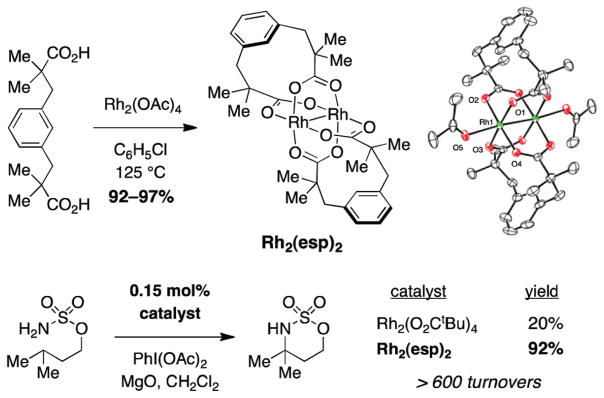
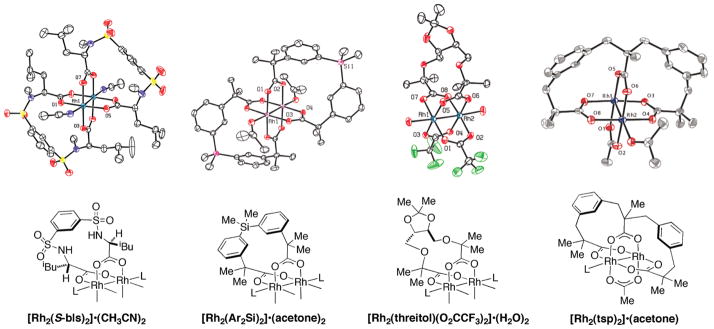
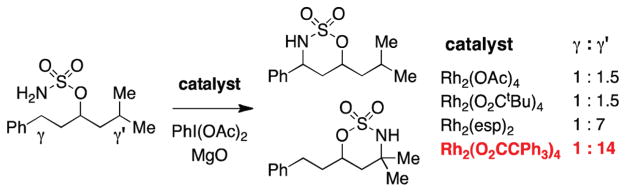
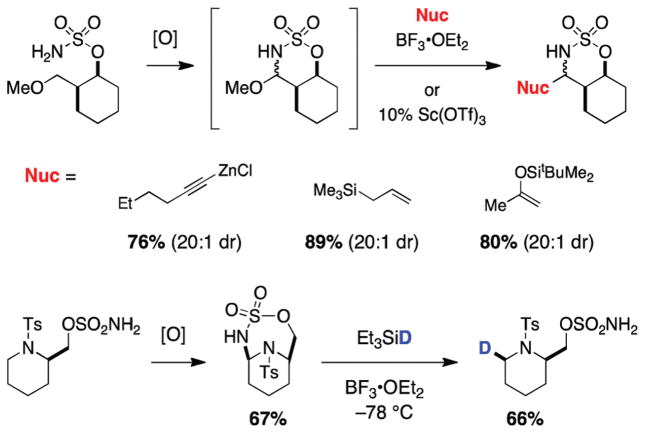
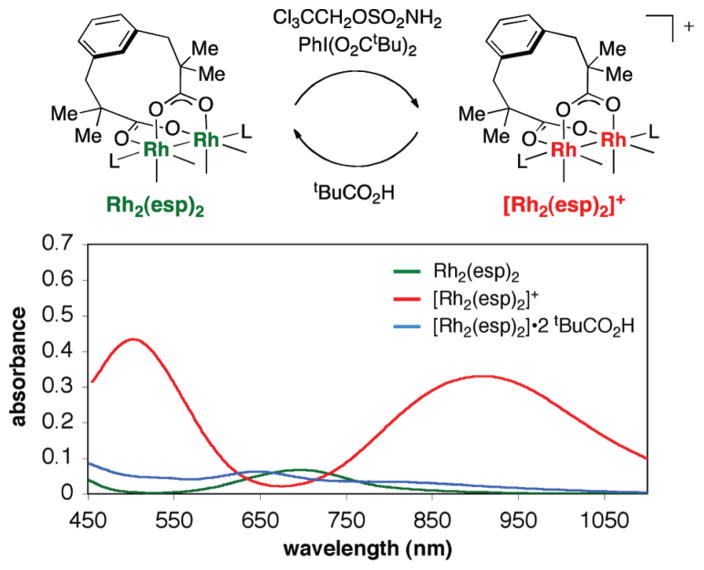
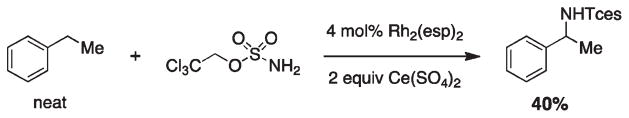
References
-
- Yu J-Q, Shi Z, editors. Topics in Current Chemistry. Vol. 292. Springer-Verlag; Berlin: 2010. C–H Activation; pp. 1–384. - PubMed
- Dyker G, editor. Handbook of C–H Transformations: Applications in Organic Synthesis. Wiley-VCH; Weinheim: 2005. pp. 1–688.
-
-
For recent reviews, see: Lebel H. In: Catalyzed Carbon-Heteroatom Bond Formation. Yudin A, editor. Wiley-VCH; Weinheim: 2011. pp. 137–156.Collet F, Dodd RH, Dauban P. Catalytic C–H amination: recent progress and future directions. Chem Commun. 2009:5061–5074.
-
-
- Masamune S. Total Syntheses of Diterpenes and Diterpene Alkaloids. IV. Garryine. J Am Chem Soc. 1964;86:290–291.
- Meyer WL, Levinson AS. Photolysis of 1,1-Dimethyl-trans-decalin-10-carbonyl Azide. Proc Chem Soc. 1963;1:15.
- ApSimon JW, Edwards OE. A New Photochemical Reaction: The Structure and Absolute Stereochemistry of Atisine. Can J Chem. 1962;40:896–902.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
