

Canalization of the evolutionary trajectory of the human influenza virus
- PMID: 22546494
- PMCID: PMC3373370
- DOI: 10.1186/1741-7007-10-38
Canalization of the evolutionary trajectory of the human influenza virus
Abstract
Background: Since its emergence in 1968, influenza A (H3N2) has evolved extensively in genotype and antigenic phenotype. However, despite strong pressure to evolve away from human immunity and to diversify in antigenic phenotype, H3N2 influenza shows paradoxically limited genetic and antigenic diversity present at any one time. Here, we propose a simple model of antigenic evolution in the influenza virus that accounts for this apparent discrepancy.
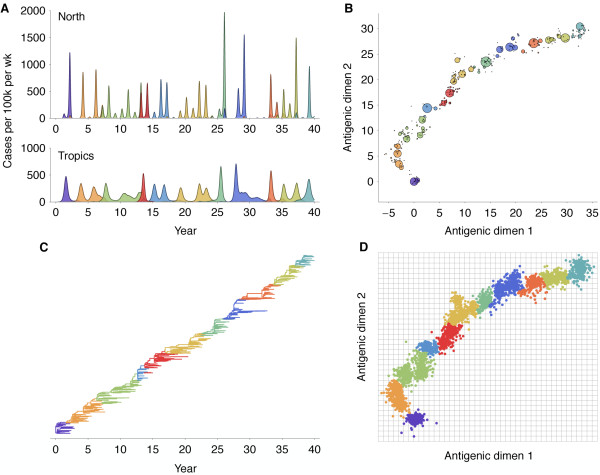
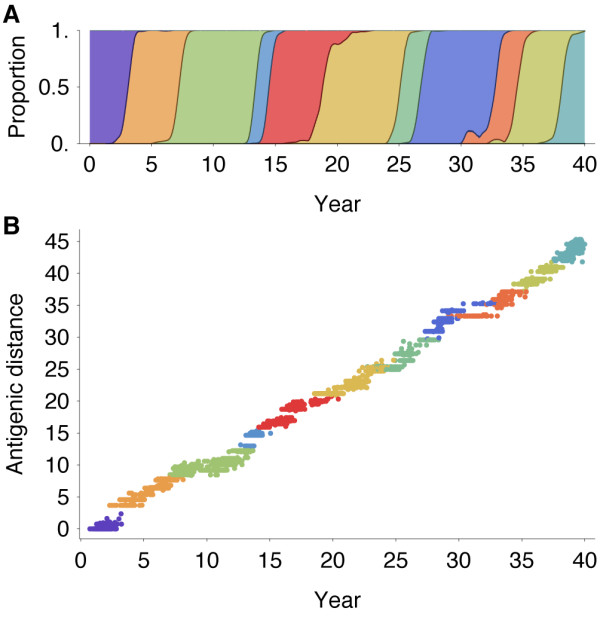
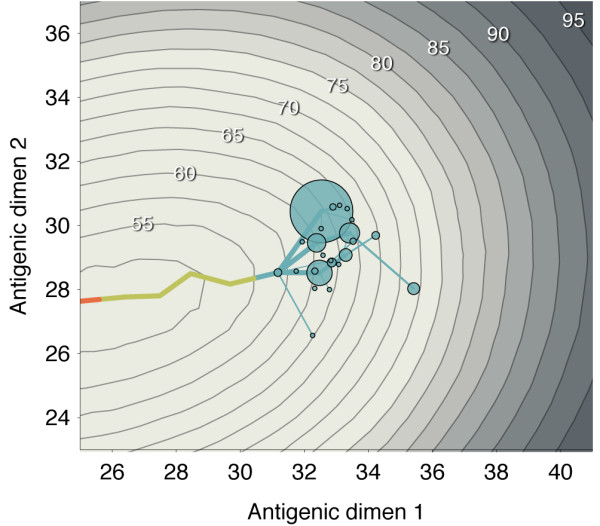
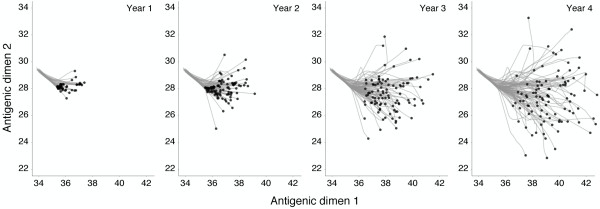
Results: In this model, antigenic phenotype is represented by a N-dimensional vector, and virus mutations perturb phenotype within this continuous Euclidean space. We implement this model in a large-scale individual-based simulation, and in doing so, we find a remarkable correspondence between model behavior and observed influenza dynamics. This model displays rapid evolution but low standing diversity and simultaneously accounts for the epidemiological, genetic, antigenic, and geographical patterns displayed by the virus. We find that evolution away from existing human immunity results in rapid population turnover in the influenza virus and that this population turnover occurs primarily along a single antigenic axis.
Conclusions: Selective dynamics induce a canalized evolutionary trajectory, in which the evolutionary fate of the influenza population is surprisingly repeatable. In the model, the influenza population shows a 1- to 2-year timescale of repeatability, suggesting a window in which evolutionary dynamics could be, in theory, predictable.
Figures

Comment in
-
Constrained evolution drives limited influenza diversity.BMC Biol. 2012 May 21;10:43. doi: 10.1186/1741-7007-10-43. BMC Biol. 2012. PMID: 22613866 Free PMC article.
References
-
- WHO. Influenza Fact Sheet. http://www.who.int/mediacentre/factsheets/fs211/en/
-
- Tria F, Lässig M, Peliti L, Franz S. A minimal stochastic model for influenza evolution. J Stat Mech. 2005. doi:10.1088/1742-5468/2005/07/P07008.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
