

Amino acid coevolution induces an evolutionary Stokes shift
- PMID: 22547823
- PMCID: PMC3361410
- DOI: 10.1073/pnas.1120084109
Amino acid coevolution induces an evolutionary Stokes shift
Abstract
The process of amino acid replacement in proteins is context-dependent, with substitution rates influenced by local structure, functional role, and amino acids at other locations. Predicting how these differences affect replacement processes is difficult. To make such inference easier, it is often assumed that the acceptabilities of different amino acids at a position are constant. However, evolutionary interactions among residue positions will tend to invalidate this assumption. Here, we use simulations of purple acid phosphatase evolution to show that amino acid propensities at a position undergo predictable change after an amino acid replacement at that position. After a replacement, the new amino acid and similar amino acids tend to become gradually more acceptable over time at that position. In other words, proteins tend to equilibrate to the presence of an amino acid at a position through replacements at other positions. Such a shift is reminiscent of the spectroscopy effect known as the Stokes shift, where molecules receiving a quantum of energy and moving to a higher electronic state will adjust to the new state and emit a smaller quantum of energy whenever they shift back down to the original ground state. Predictions of changes in stability in real proteins show that mutation reversals become less favorable over time, and thus, broadly support our results. The observation of an evolutionary Stokes shift has profound implications for the study of protein evolution and the modeling of evolutionary processes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
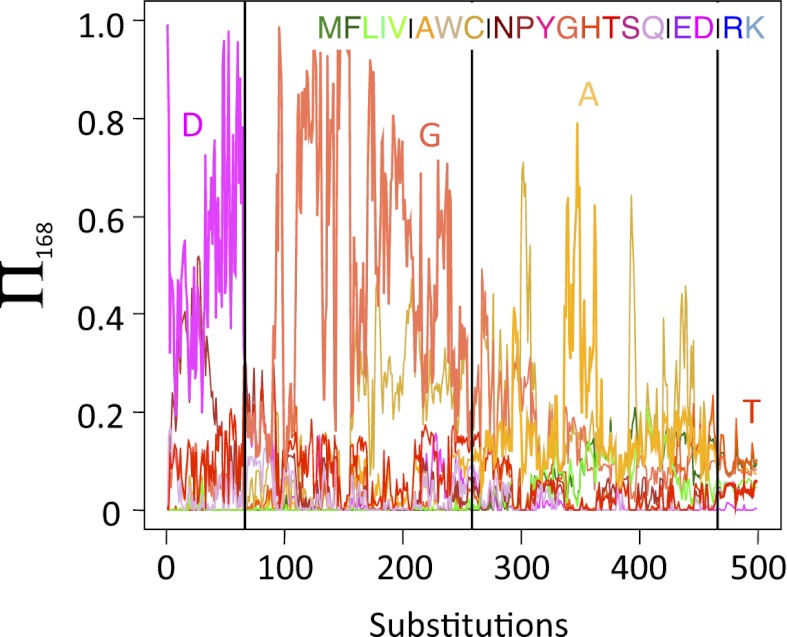


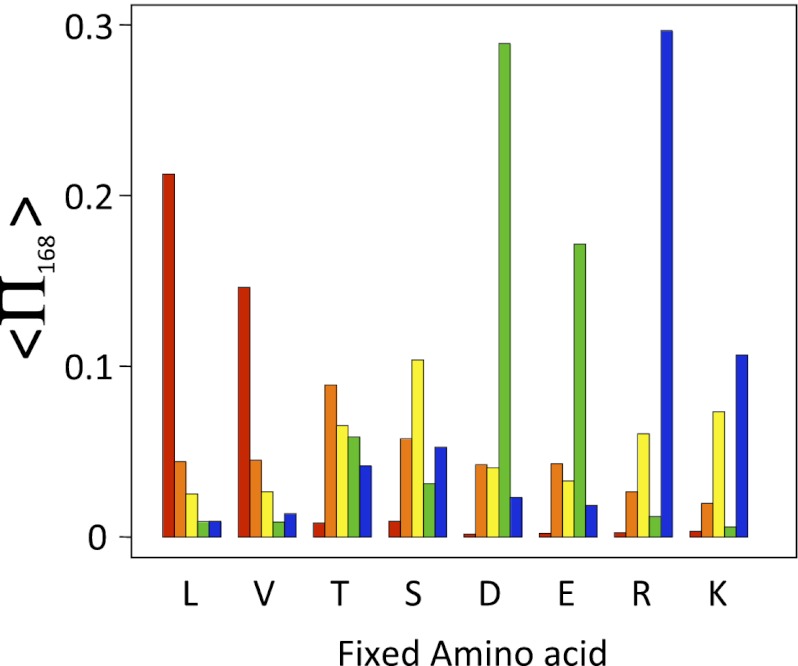
 ) depending on the amino acid fixed at that location. Results are shown for X equal to lysine (L; red), threonine (T; orange), serine (S; yellow), aspartic acid (D; green), and arginine (R; blue).
) depending on the amino acid fixed at that location. Results are shown for X equal to lysine (L; red), threonine (T; orange), serine (S; yellow), aspartic acid (D; green), and arginine (R; blue).
 ) after an R110D mutation. Three sequences were chosen for which an R111D mutation was slightly deleterious (
) after an R110D mutation. Three sequences were chosen for which an R111D mutation was slightly deleterious ( ). This mutation was made, and the D was fixed at this location. Red, blue, and green traces represent individual simulations. Black curve represents the average of 1,000 simulations for each of the three initial sequences.
). This mutation was made, and the D was fixed at this location. Red, blue, and green traces represent individual simulations. Black curve represents the average of 1,000 simulations for each of the three initial sequences.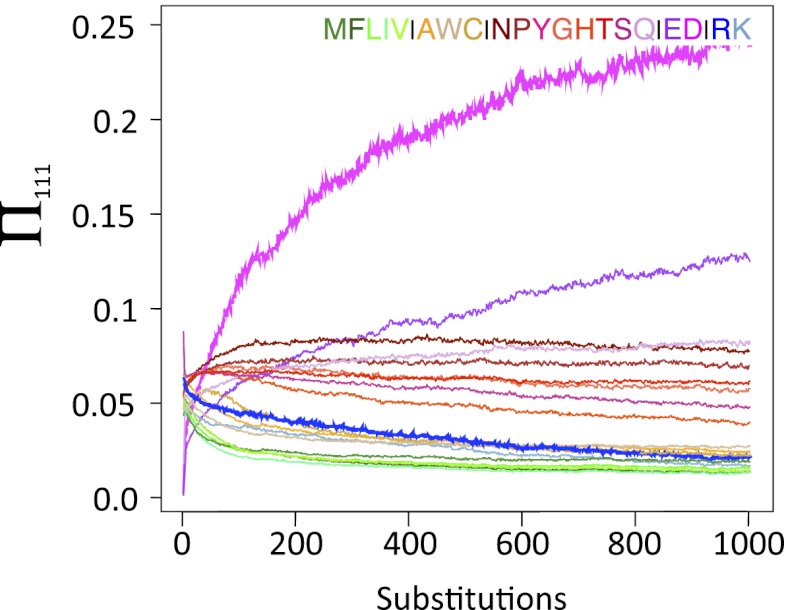
 ). This mutation was made, and the D was fixed at this location. Curves represent the average of 1,000 simulations for each of the three initial sequences. Amino acid color codes are as in Figs. 1 and 2.
). This mutation was made, and the D was fixed at this location. Curves represent the average of 1,000 simulations for each of the three initial sequences. Amino acid color codes are as in Figs. 1 and 2.
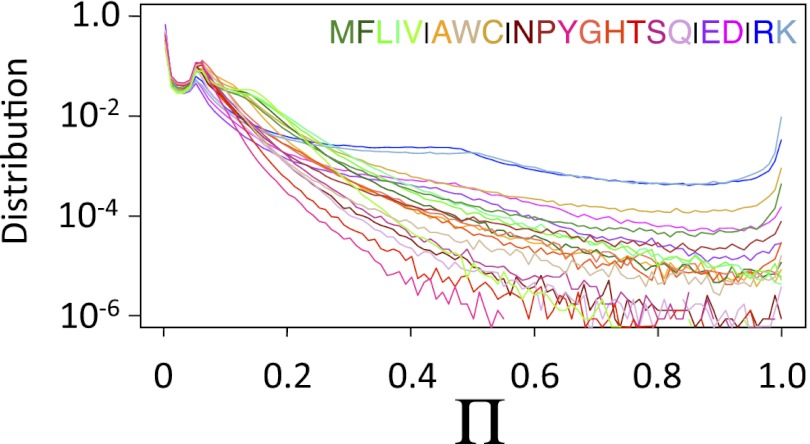
 compared with
compared with  for mutations where one sequence is changed to match the amino acid in the other sequence at that location as a function of the pairwise identity at other locations: (A) 100%, (C) 75%, and (E) 20%. If all locations are independent,
for mutations where one sequence is changed to match the amino acid in the other sequence at that location as a function of the pairwise identity at other locations: (A) 100%, (C) 75%, and (E) 20%. If all locations are independent,  , which is the case for A. (B, D, and F) Similar calculations for different homologs of ferrodoxin, where the values of
, which is the case for A. (B, D, and F) Similar calculations for different homologs of ferrodoxin, where the values of  and
and  are computed using Rosetta (31). Calculations were based on the crystal structures of the two proteins for sets of proteins where the pairwise identity at other locations was (B) 100%, (D) 70–80%, or (E) <25%. Correlation coefficients (cc), calculated after excluding outliers (
are computed using Rosetta (31). Calculations were based on the crystal structures of the two proteins for sets of proteins where the pairwise identity at other locations was (B) 100%, (D) 70–80%, or (E) <25%. Correlation coefficients (cc), calculated after excluding outliers ( ), are included in the plots.
), are included in the plots.
References
-
- Pollock DD, Bruno WJ. Assessing an unknown evolutionary process: Effect of increasing site-specific knowledge through taxon addition. Mol Biol Evol. 2000;17:1854–1858. - PubMed
-
- Dimmic MW, Hubisz MJ, Bustamante CD, Nielsen R. Detecting coevolving amino acid sites using Bayesian mutational mapping. Bioinformatics. 2005;21(Suppl 1):i126–i135. - PubMed
-
- Göbel U, Sander C, Schneider R, Valencia A. Correlated mutations and residue contacts in proteins. Proteins. 1994;18:309–317. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
