

MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors
- PMID: 22552284
- PMCID: PMC3515079
- DOI: 10.1158/0008-5472.CAN-11-3747
MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors
Abstract
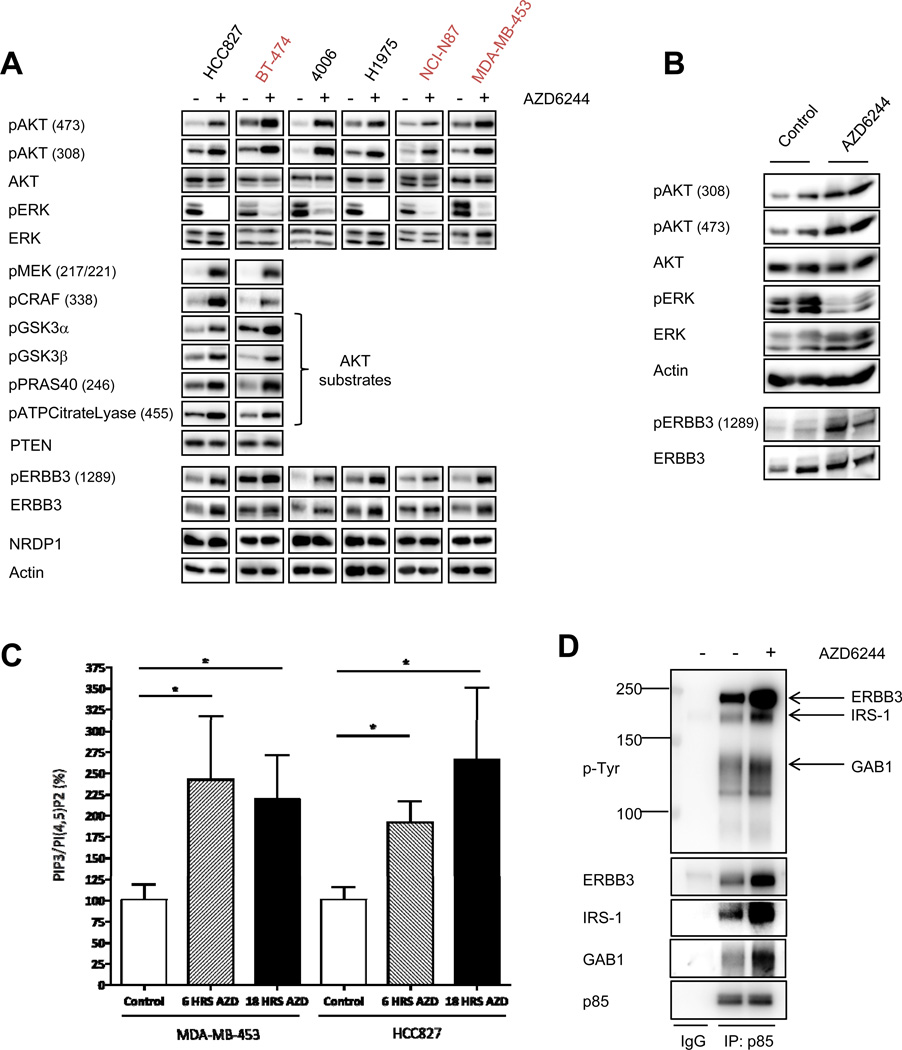
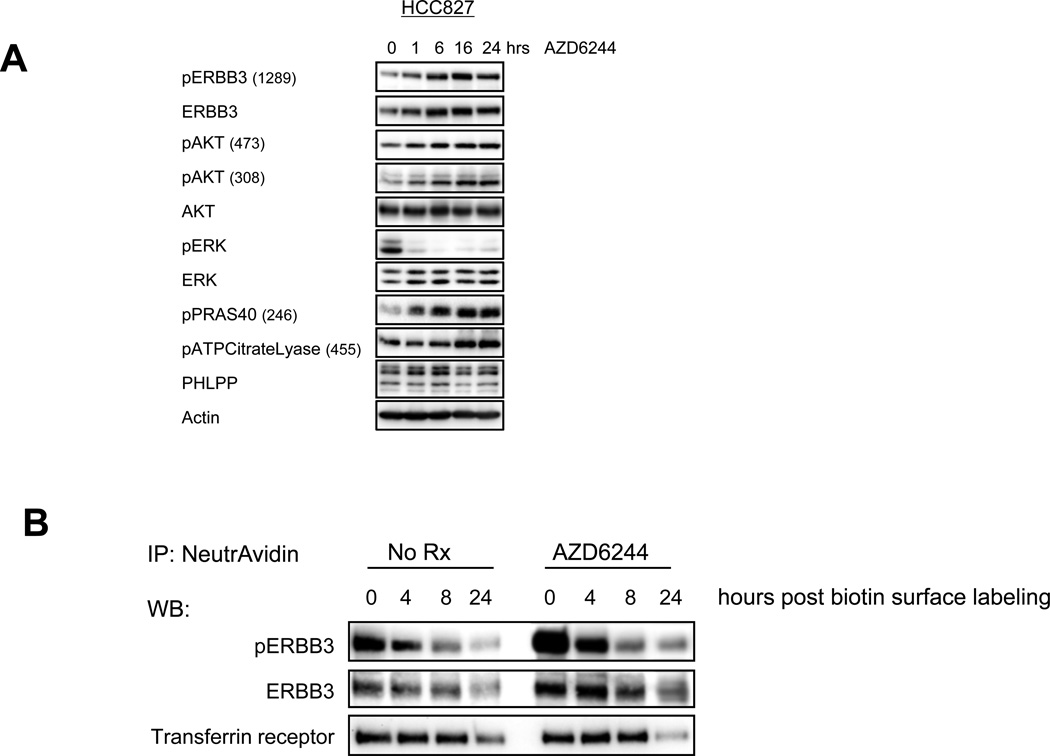
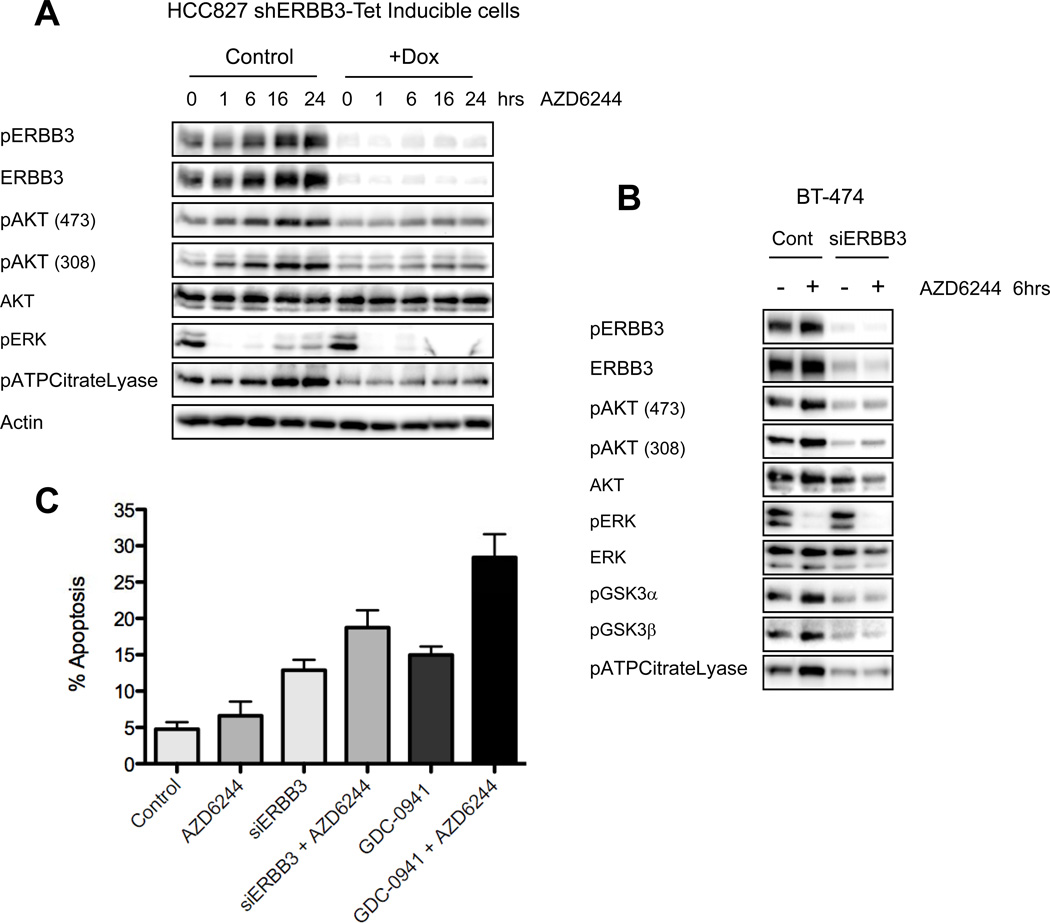
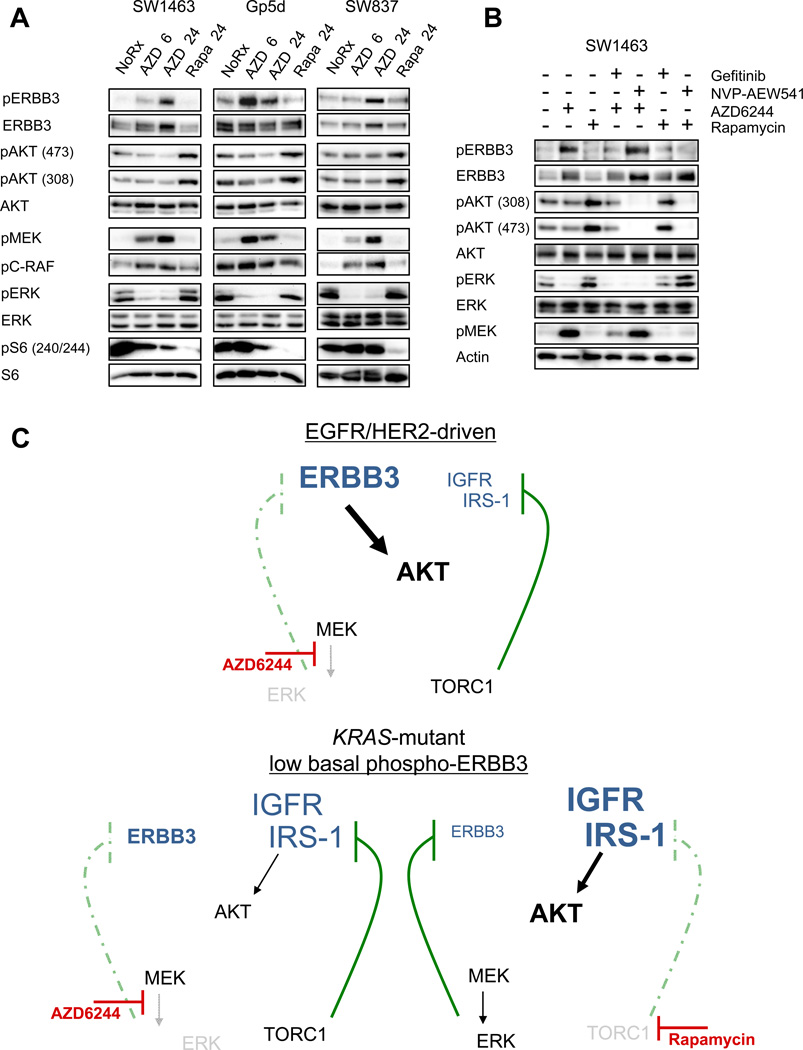
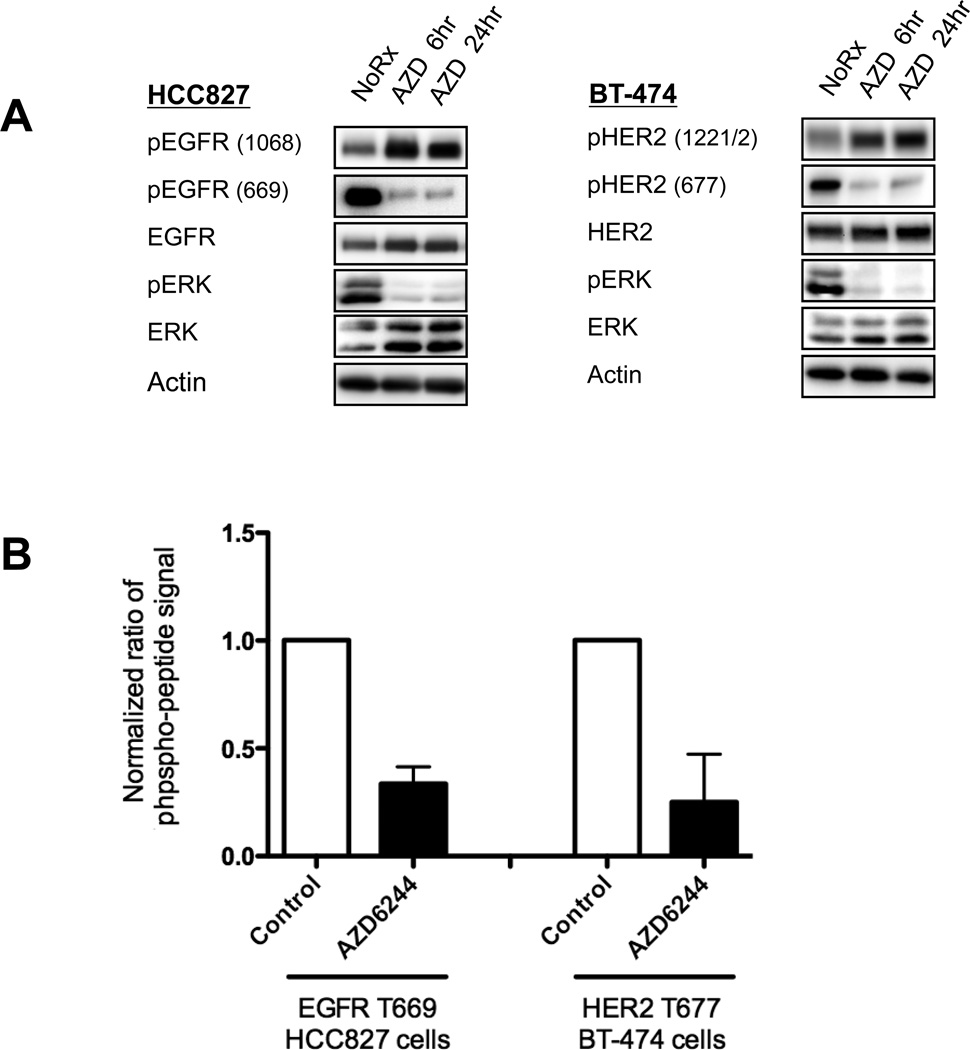
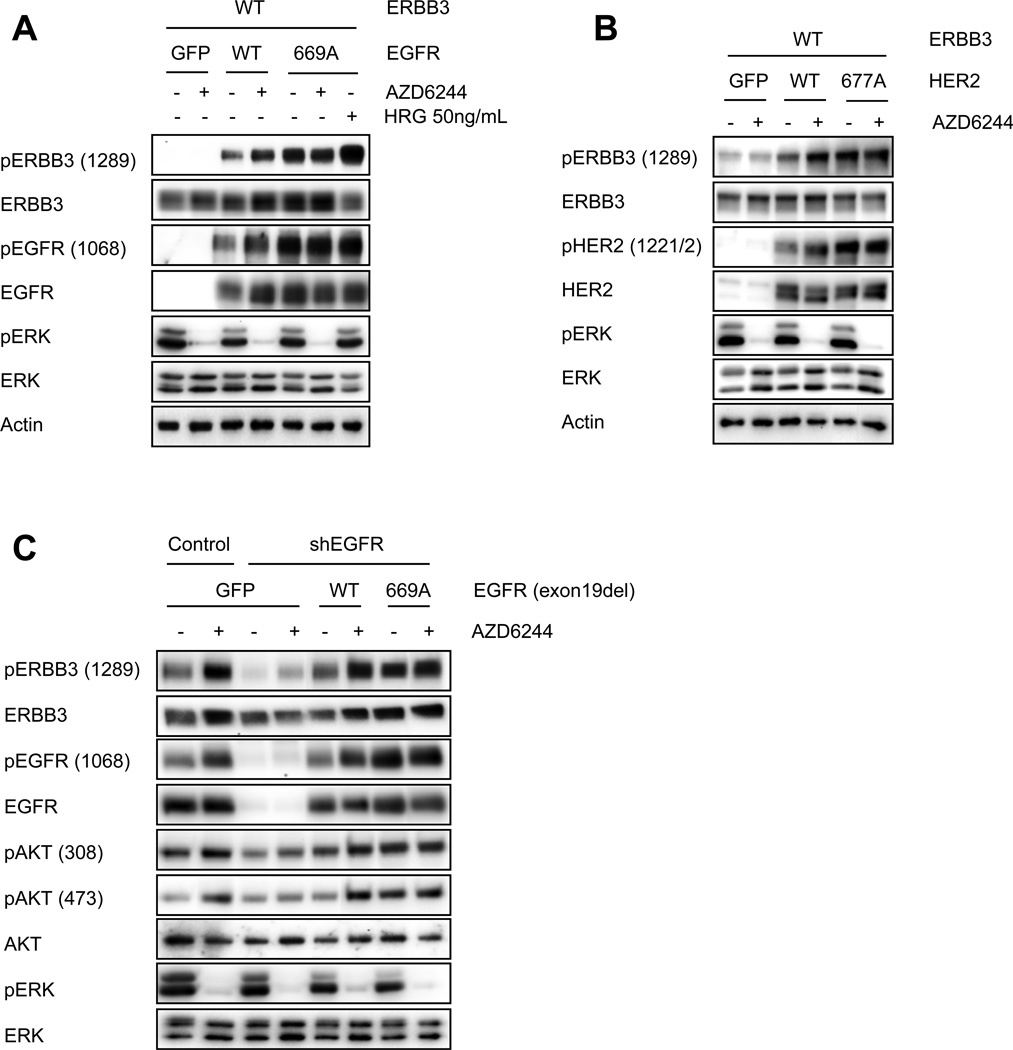
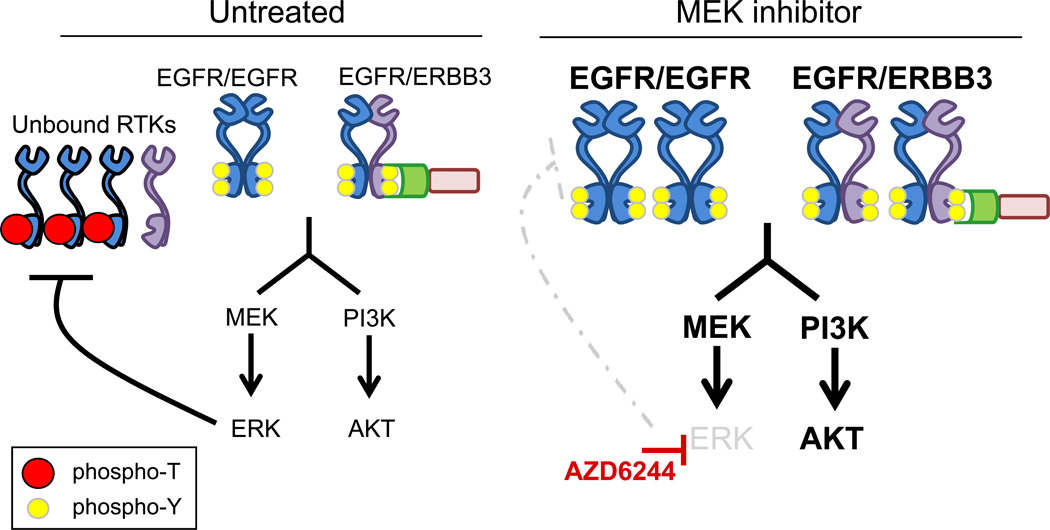
The phosphoinositide 3-kinase (PI3K)/AKT and RAF/MEK/ERK signaling pathways are activated in a wide range of human cancers. In many cases, concomitant inhibition of both pathways is necessary to block proliferation and induce cell death and tumor shrinkage. Several feedback systems have been described in which inhibition of one intracellular pathway leads to activation of a parallel signaling pathway, thereby decreasing the effectiveness of single-agent targeted therapies. In this study, we describe a feedback mechanism in which MEK inhibition leads to activation of PI3K/AKT signaling in EGFR and HER2-driven cancers. We found that MEK inhibitor-induced activation of PI3K/AKT resulted from hyperactivation of ERBB3 as a result of the loss of an inhibitory threonine phosphorylation in the conserved juxtamembrane domains of EGFR and HER2. Mutation of this amino acid led to increased ERBB receptor activation and upregulation of the ERBB3/PI3K/AKT signaling pathway, which was no longer responsive to MEK inhibition. Taken together, these results elucidate an important, dominant feedback network regulating central oncogenic pathways in human cancer.
©2012 AACR.
Conflict of interest statement
Figures
References
-
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. - PubMed
-
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. - PubMed
-
- Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009;283:125–134. - PubMed
-
- Sun S, LM R, X W, Z Z, P Y, H F, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Research. 2005;65:7052–7058. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA080195/CA/NCI NIH HHS/United States
- P50CA090578/CA/NCI NIH HHS/United States
- P50 CA98131/CA/NCI NIH HHS/United States
- P01 CA120964/CA/NCI NIH HHS/United States
- 5P01CA120964-04/CA/NCI NIH HHS/United States
- 1U01CA141457-01/CA/NCI NIH HHS/United States
- P50 CA098131/CA/NCI NIH HHS/United States
- R01 CA140594/CA/NCI NIH HHS/United States
- P50 CA090578/CA/NCI NIH HHS/United States
- K08 CA120060-01/CA/NCI NIH HHS/United States
- P50 CA127003/CA/NCI NIH HHS/United States
- P30 CA006516/CA/NCI NIH HHS/United States
- R01CA80195/CA/NCI NIH HHS/United States
- R01CA140594/CA/NCI NIH HHS/United States
- R01 CA137008/CA/NCI NIH HHS/United States
- U01 CA141457/CA/NCI NIH HHS/United States
- R01 CA166480/CA/NCI NIH HHS/United States
- K08 CA120060/CA/NCI NIH HHS/United States
- R01CA137008-01/CA/NCI NIH HHS/United States
- 5P30CA006516-46/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
