

Modeling microbial community structure and functional diversity across time and space
- PMID: 22553907
- PMCID: PMC3396557
- DOI: 10.1111/j.1574-6968.2012.02588.x
Modeling microbial community structure and functional diversity across time and space
Abstract
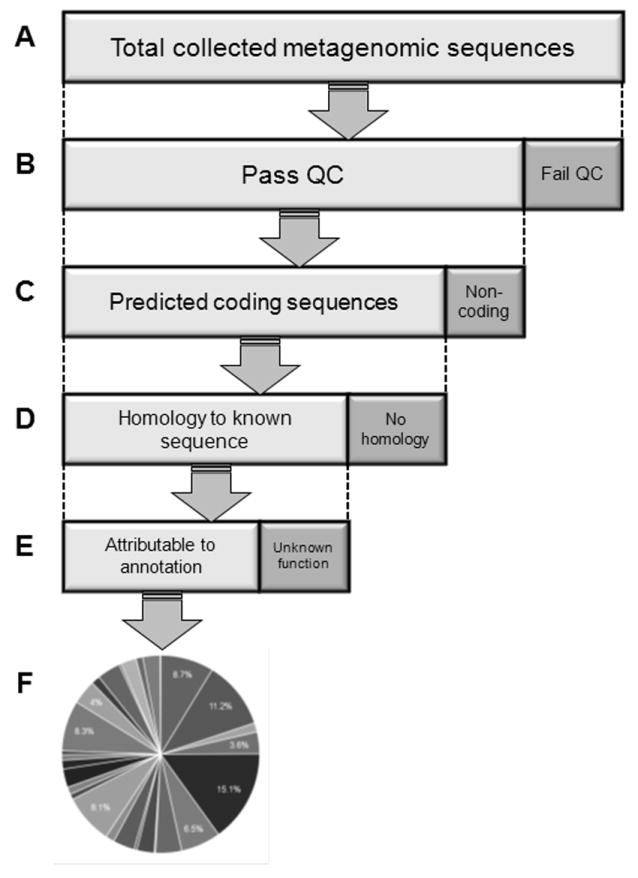
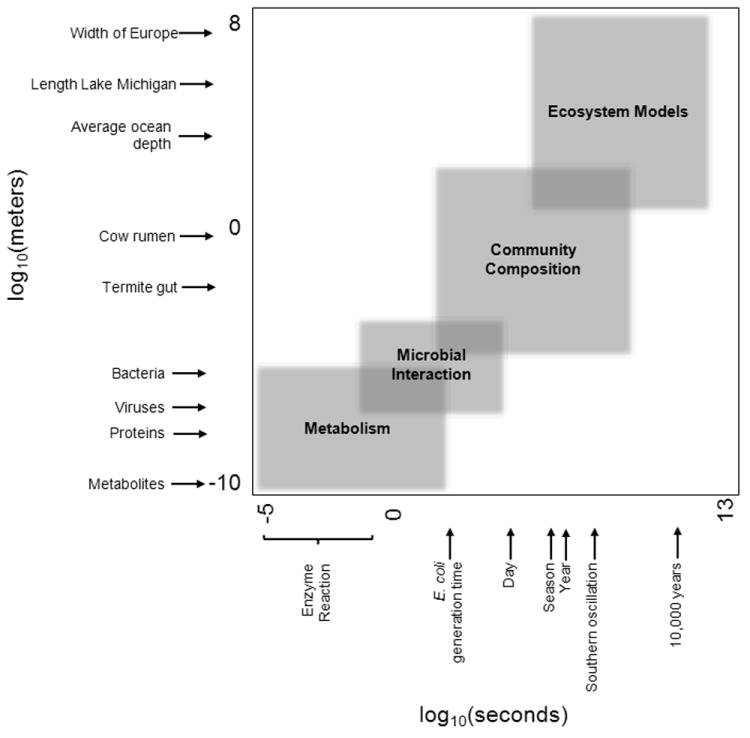
Microbial communities exhibit exquisitely complex structure. Many aspects of this complexity, from the number of species to the total number of interactions, are currently very difficult to examine directly. However, extraordinary efforts are being made to make these systems accessible to scientific investigation. While recent advances in high-throughput sequencing technologies have improved accessibility to the taxonomic and functional diversity of complex communities, monitoring the dynamics of these systems over time and space - using appropriate experimental design - is still expensive. Fortunately, modeling can be used as a lens to focus low-resolution observations of community dynamics to enable mathematical abstractions of functional and taxonomic dynamics across space and time. Here, we review the approaches for modeling bacterial diversity at both the very large and the very small scales at which microbial systems interact with their environments. We show that modeling can help to connect biogeochemical processes to specific microbial metabolic pathways.
© 2012 Federation of European Microbiological Societies. Published by Blackwell Publishing Ltd. All rights reserved.
Figures
References
-
- Bardgett RD, Freeman C, Ostle NJ. Microbial contributions to climate change through carbon cycle feedbacks. ISME J. 2008;2:805–814. - PubMed
-
- Baune C, Bottcher ME. Experimental investigation of sulphur isotope partitioning during outgassing of hydrogen sulphide from diluted aqueous solutions and seawater. Isotopes in Env and Health Stud. 2010;46:444–453. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
