

Stabilizing proteins from sequence statistics: the interplay of conservation and correlation in triosephosphate isomerase stability
- PMID: 22555051
- PMCID: PMC3578383
- DOI: 10.1016/j.jmb.2012.04.025
Stabilizing proteins from sequence statistics: the interplay of conservation and correlation in triosephosphate isomerase stability
Abstract
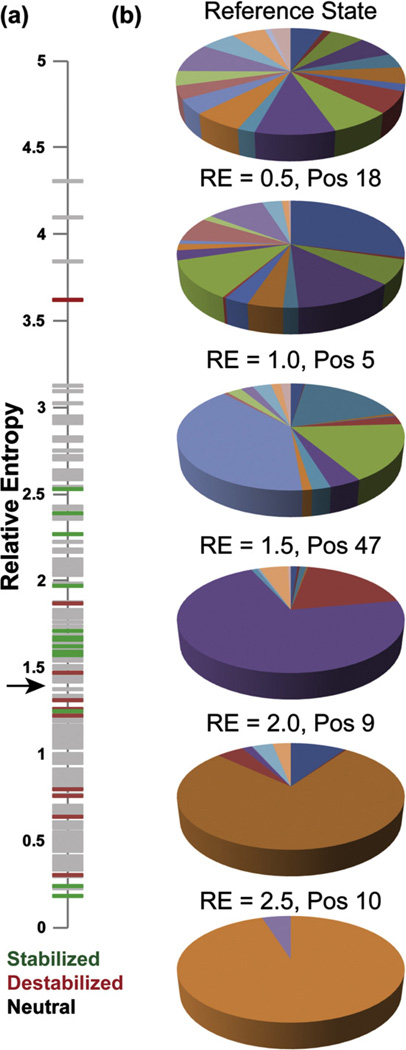
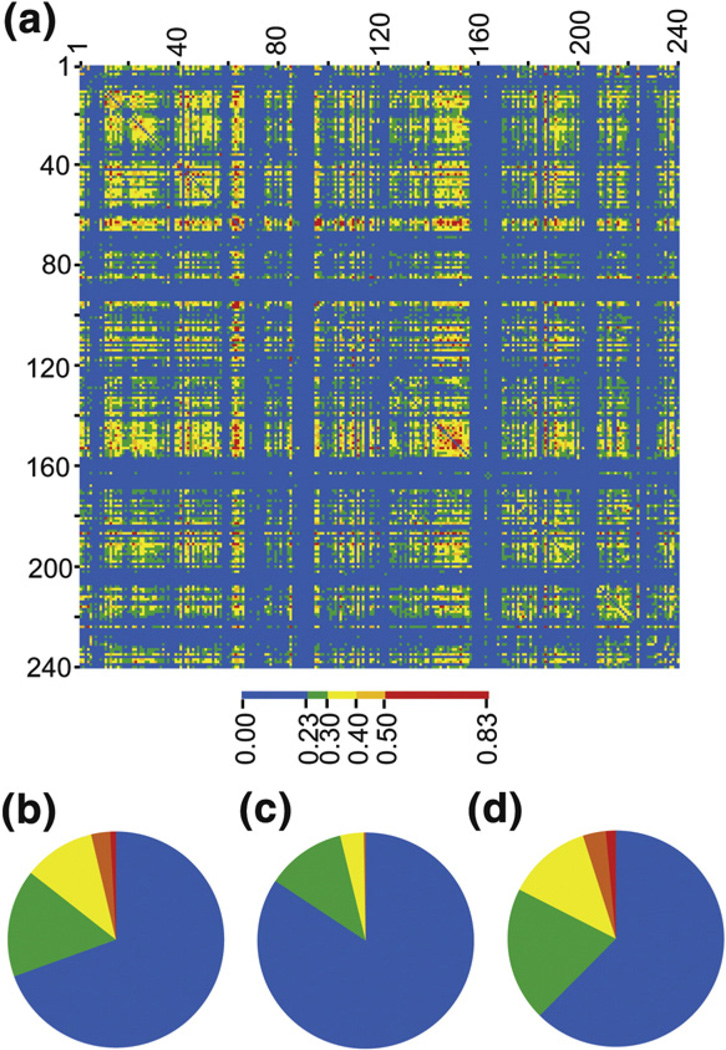
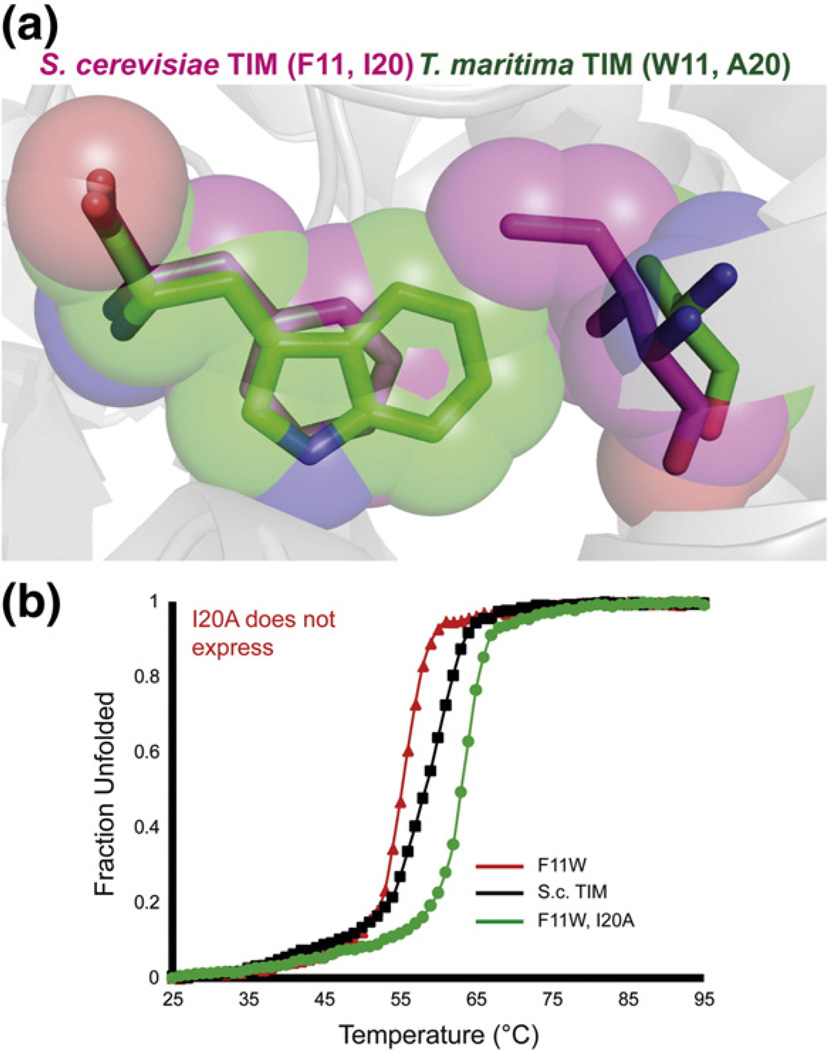
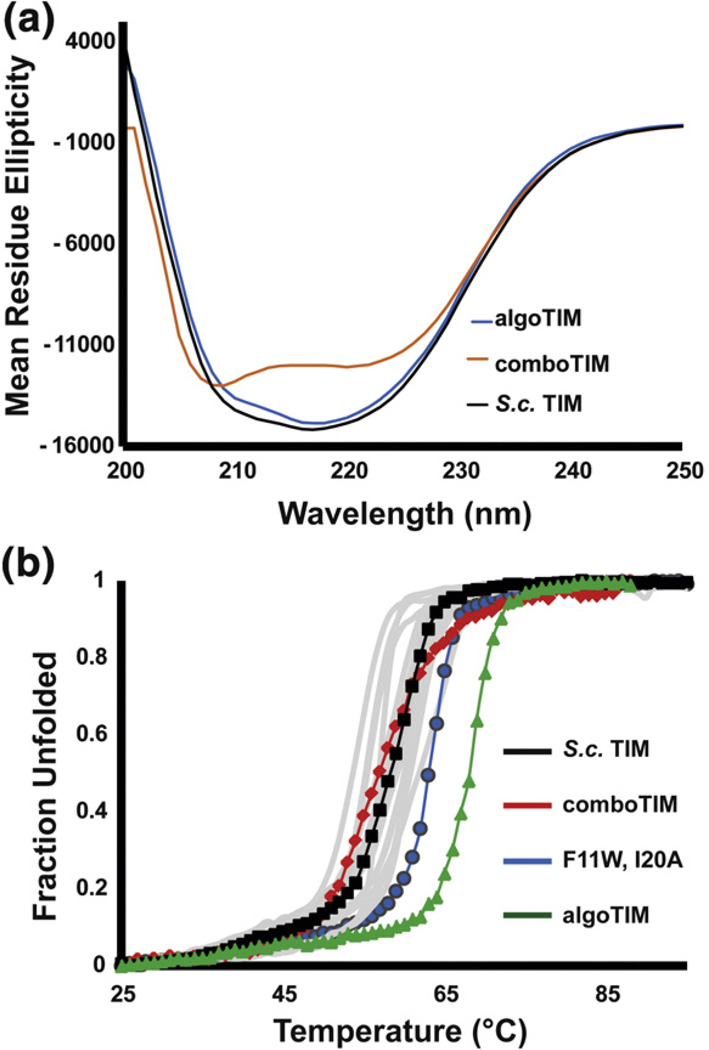
Understanding the determinants of protein stability remains one of protein science's greatest challenges. There are still no computational solutions that calculate the stability effects of even point mutations with sufficient reliability for practical use. Amino acid substitutions rarely increase the stability of native proteins; hence, large libraries and high-throughput screens or selections are needed to stabilize proteins using directed evolution. Consensus mutations have proven effective for increasing stability, but these mutations are successful only about half the time. We set out to understand why some consensus mutations fail to stabilize, and what criteria might be useful to predict stabilization more accurately. Overall, consensus mutations at more conserved positions were more likely to be stabilizing in our model, triosephosphate isomerase (TIM) from Saccharomyces cerevisiae. However, positions coupled to other sites were more likely not to stabilize upon mutation. Destabilizing mutations could be removed both by removing sites with high statistical correlations to other positions and by removing nearly invariant positions at which "hidden correlations" can occur. Application of these rules resulted in identification of stabilizing mutations in 9 out of 10 positions, and amalgamation of all predicted stabilizing positions resulted in the most stable yeast TIM variant we produced (+8 °C). In contrast, a multimutant with 14 mutations each found to stabilize TIM independently was destabilized by 2 °C. Our results are a practical extension to the consensus concept of protein stabilization, and they further suggest the importance of positional independence in the mechanism of consensus stabilization.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures
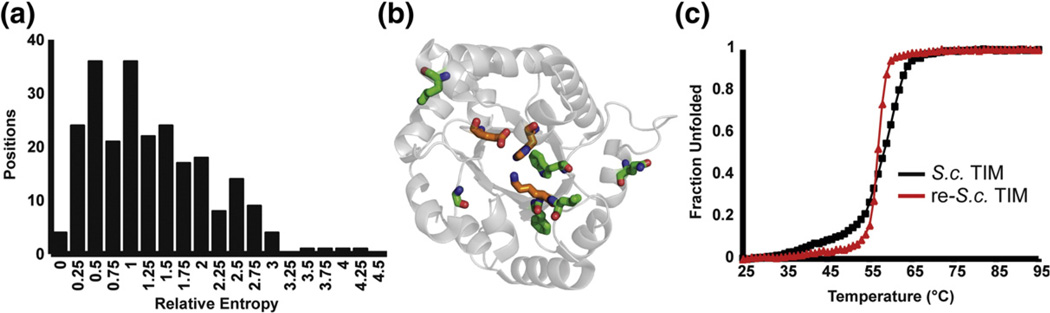
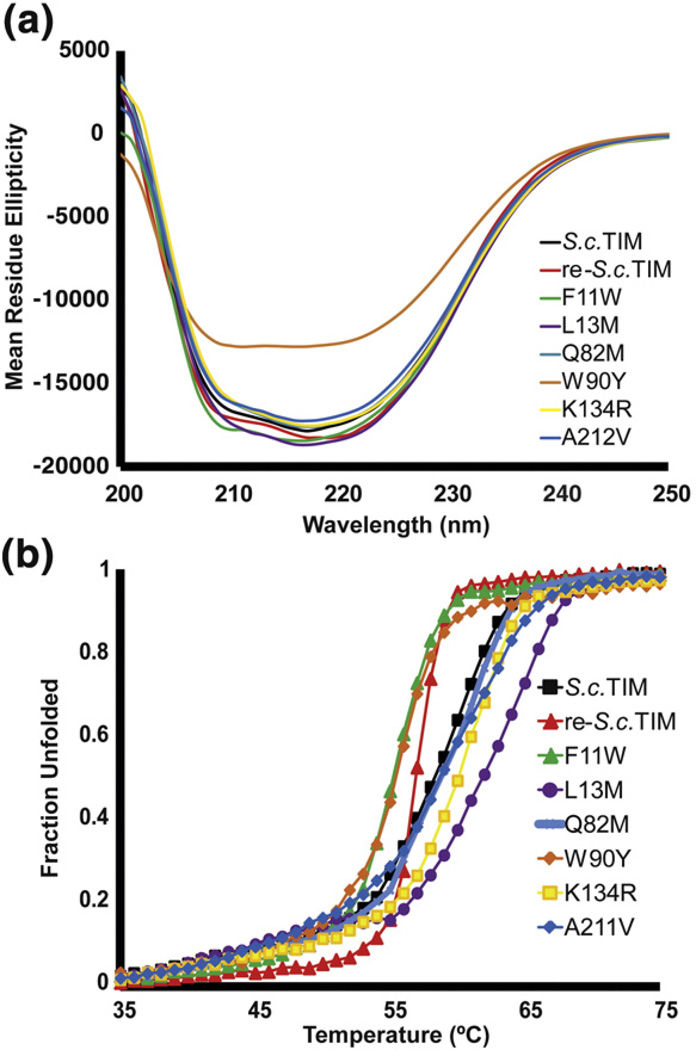
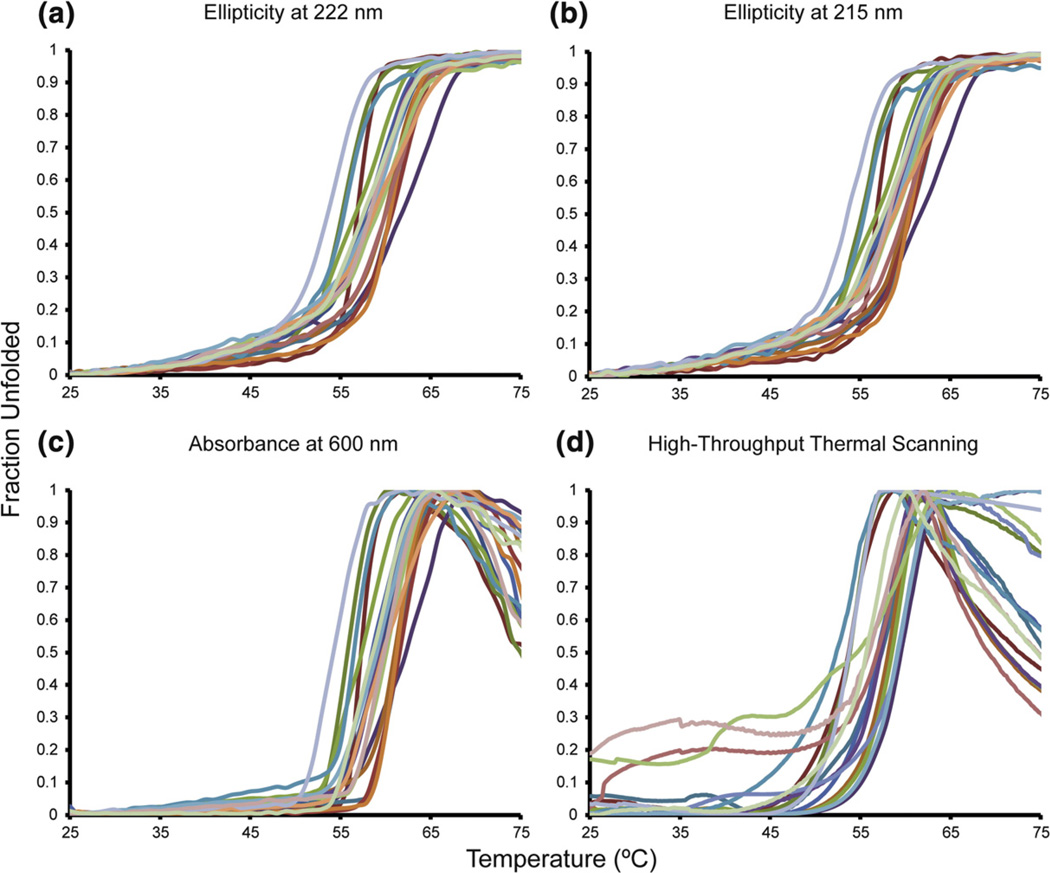
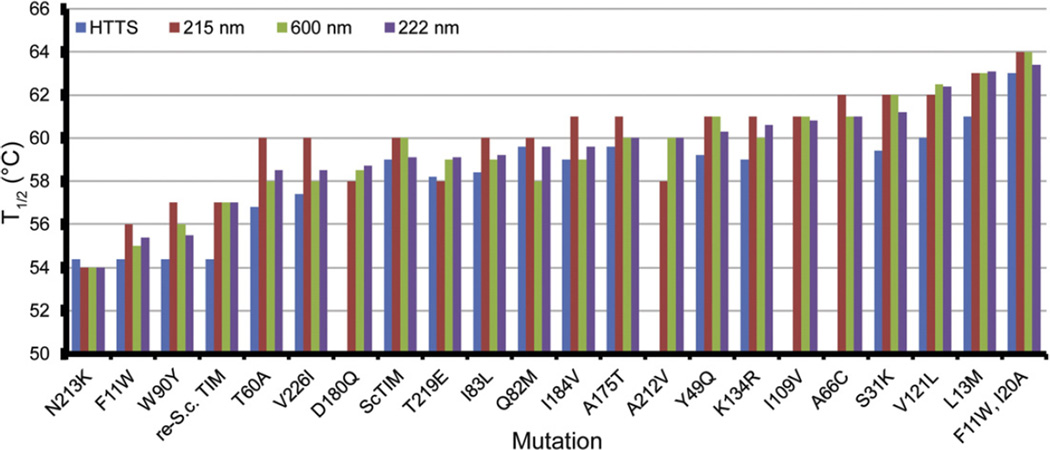
References
-
- Dill KA. Dominant forces in protein folding. Biochemistry. 1990;29:7133–7155. - PubMed
-
- Rose GD, Wolfenden R. Hydrogen bonding, hydrophobicity, packing, and protein folding. Annu. Rev. Biophys. Biomol. Struct. 1993;22:381–415. - PubMed
-
- Cordes MH, Davidson AR, Sauer RT. Sequence space, folding and protein design. Curr. Opin. Struct. Biol. 1996;6:3–10. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
