

Genetic control of canine leishmaniasis: genome-wide association study and genomic selection analysis
- PMID: 22558142
- PMCID: PMC3338836
- DOI: 10.1371/journal.pone.0035349
Genetic control of canine leishmaniasis: genome-wide association study and genomic selection analysis
Abstract
Background: The current disease model for leishmaniasis suggests that only a proportion of infected individuals develop clinical disease, while others are asymptomatically infected due to immune control of infection. The factors that determine whether individuals progress to clinical disease following Leishmania infection are unclear, although previous studies suggest a role for host genetics. Our hypothesis was that canine leishmaniasis is a complex disease with multiple loci responsible for the progression of the disease from Leishmania infection.
Methodology/principal findings: Genome-wide association and genomic selection approaches were applied to a population-based case-control dataset of 219 dogs from a single breed (Boxer) genotyped for ~170,000 SNPs. Firstly, we aimed to identify individual disease loci; secondly, we quantified the genetic component of the observed phenotypic variance; and thirdly, we tested whether genome-wide SNP data could accurately predict the disease.
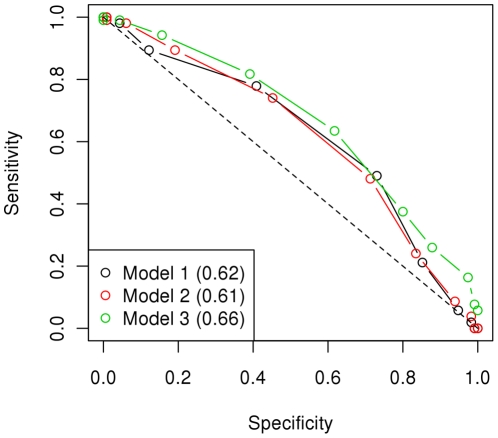
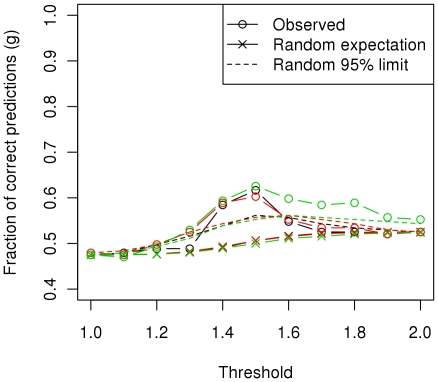
Conclusions/significance: We estimated that a substantial proportion of the genome is affecting the trait and that its heritability could be as high as 60%. Using the genome-wide association approach, the strongest associations were on chromosomes 1, 4 and 20, although none of these were statistically significant at a genome-wide level and after correcting for genetic stratification and lifestyle. Amongst these associations, chromosome 4: 61.2-76.9 Mb maps to a locus that has previously been associated with host susceptibility to human and murine leishmaniasis, and genomic selection estimated markers in this region to have the greatest effect on the phenotype. We therefore propose these regions as candidates for replication studies. An important finding of this study was the significant predictive value from using the genomic information. We found that the phenotype could be predicted with an accuracy of ~0.29 in new samples and that the affection status was correctly predicted in 60% of dogs, significantly higher than expected by chance, and with satisfactory sensitivity-specificity values (AUC = 0.63).
Conflict of interest statement
Figures
References
-
- Blackwell JM. Genetic susceptibility to leishmanial infections: studies in mice and man. Parasitology. 1996;112(Supplement S1):S67. - PubMed
-
- Ibrahim M, Lambson B, Yousif A, Deifalla N, Alnaiem D, et al. Kala-azar in a high transmission focus: an ethnic and geographic dimension. Am J Trop Med Hyg. 1999;61(6):941–944. - PubMed
-
- Cabello PH, Lima AMVMD, Azevedo ES, Krieger H. Familial Aggregation of Leishmania chagasi Infection in Northeastern Brazil. Am J Trop Med Hyg. 1995;52(4):364–365. - PubMed
-
- Zijlstra EE, El-Hassan AM, Ismael A, Ghalib HW. Endemic Kala-Azar in Eastern Sudan: A Longitudinal Study on the Incidence of Clinical and Subclinical Infection and Post-Kala-Azar Dermal Leishmaniasis,. Am J Trop Med Hyg. 1994;51,(6,):826–836. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- G0900753/MRC_/Medical Research Council/United Kingdom
- BBS/E/D/05191132/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- G0600237/MRC_/Medical Research Council/United Kingdom
- G0100594/MRC_/Medical Research Council/United Kingdom
- G0901461/MRC_/Medical Research Council/United Kingdom
