

Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation
- PMID: 22562073
- PMCID: PMC3429541
- DOI: 10.4161/auto.20123
Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation
Abstract
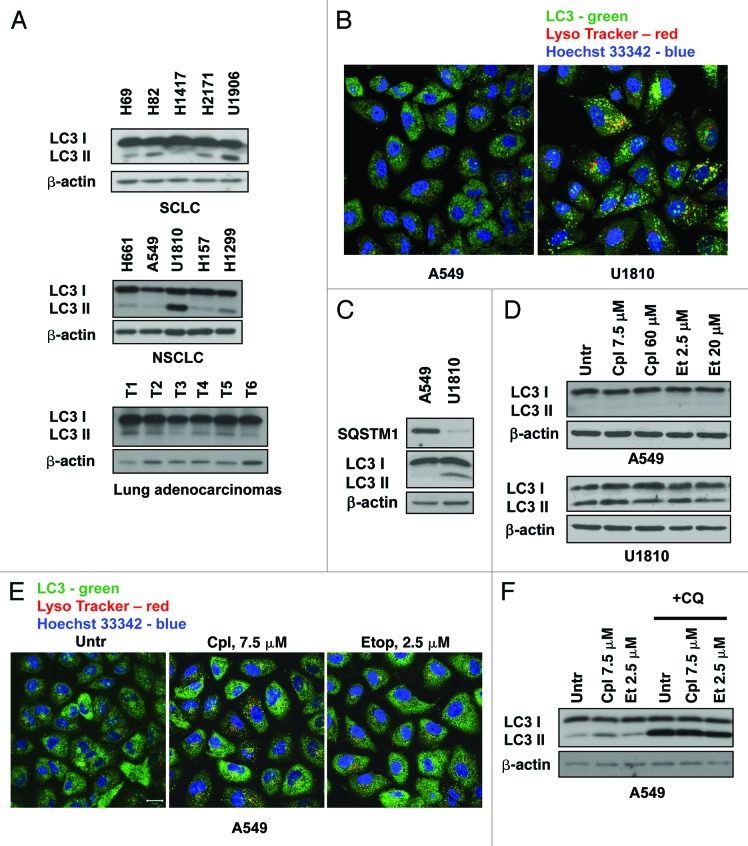
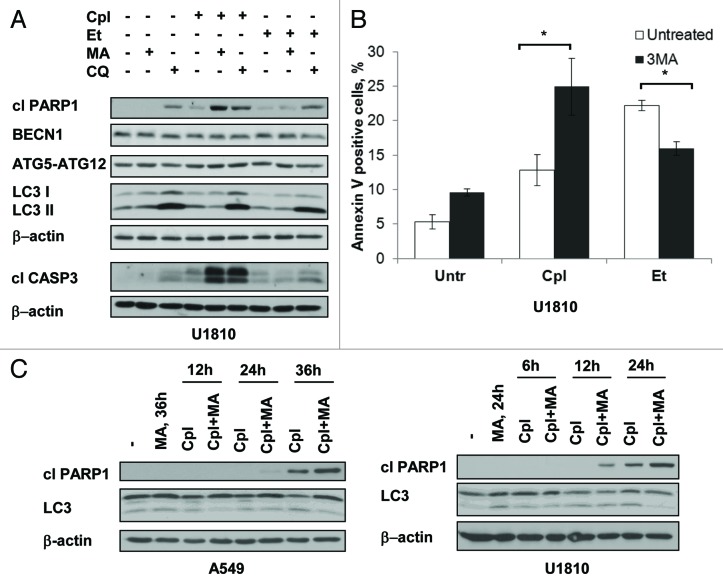
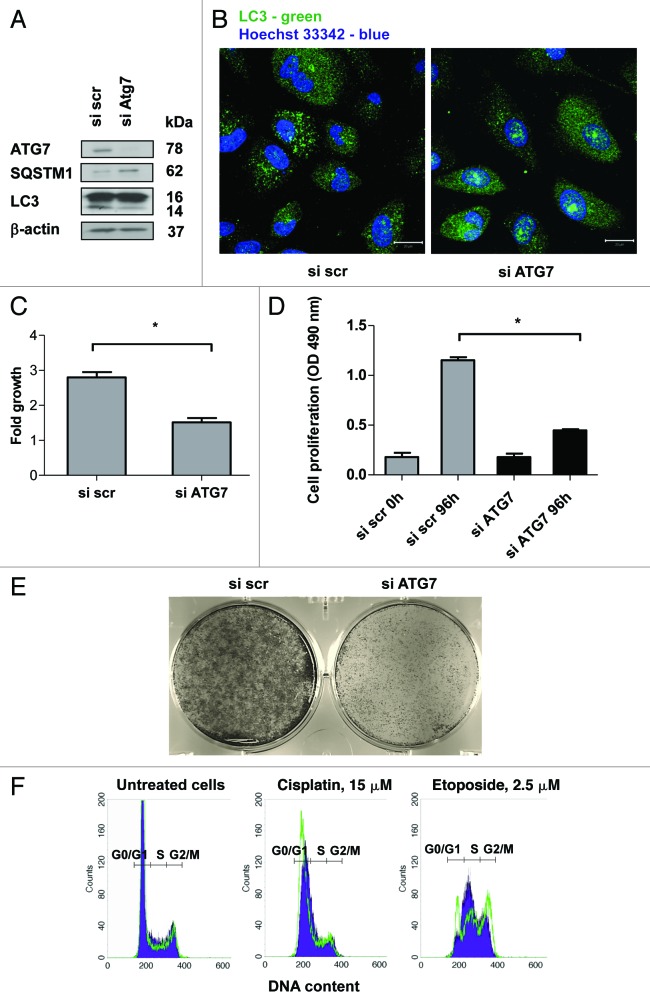
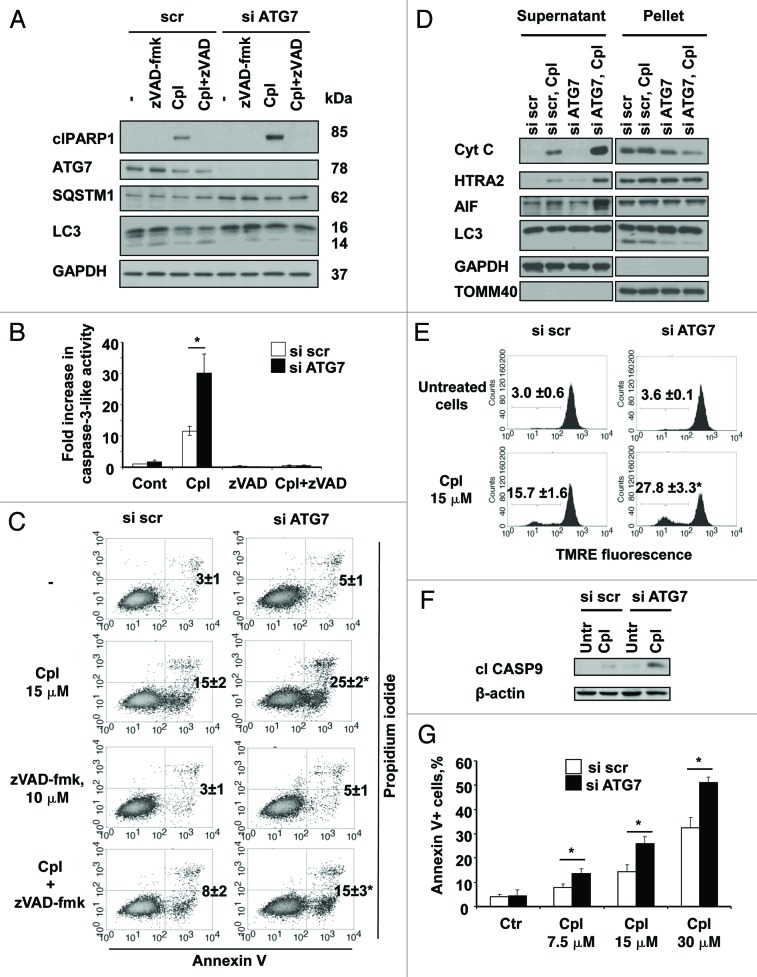
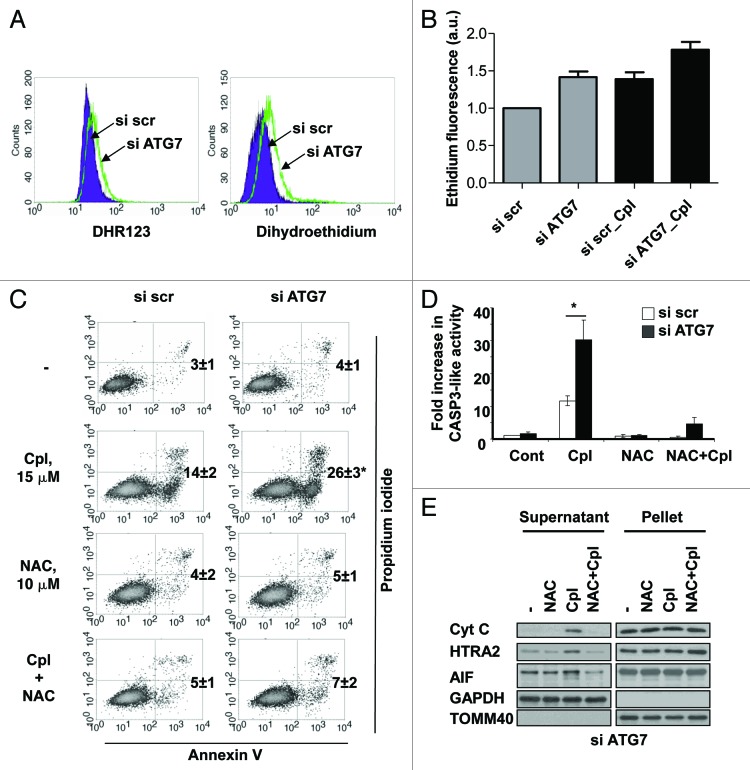
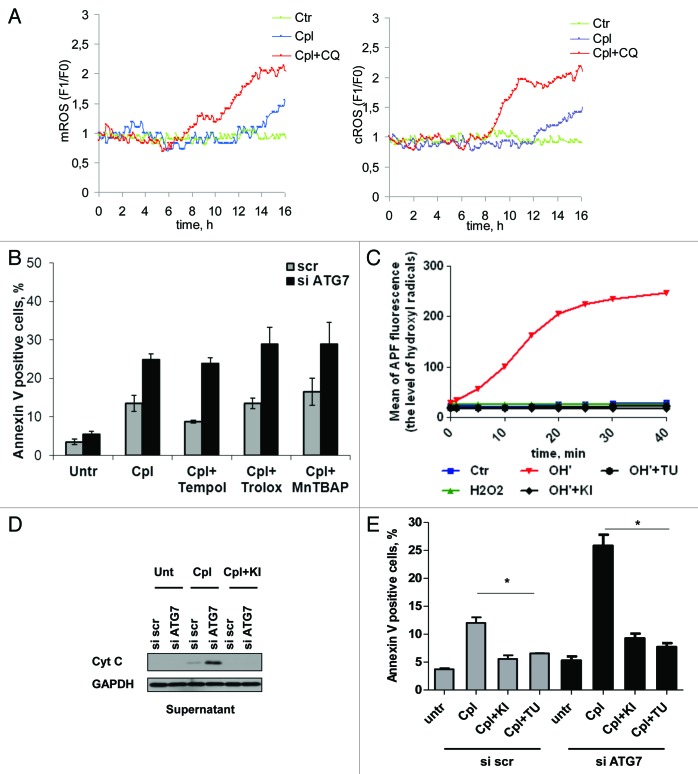
Autophagy is a catabolic process involved in the turnover of organelles and macromolecules which, depending on conditions, may lead to cell death or preserve cell survival. We found that some lung cancer cell lines and tumor samples are characterized by increased levels of lipidated LC3. Inhibition of autophagy sensitized non-small cell lung carcinoma (NSCLC) cells to cisplatin-induced apoptosis; however, such response was attenuated in cells treated with etoposide. Inhibition of autophagy stimulated ROS formation and treatment with cisplatin had a synergistic effect on ROS accumulation. Using genetically encoded hydrogen peroxide probes directed to intracellular compartments we found that autophagy inhibition facilitated formation of hydrogen peroxide in the cytosol and mitochondria of cisplatin-treated cells. The enhancement of cell death under conditions of inhibited autophagy was partially dependent on caspases, however, antioxidant NAC or hydroxyl radical scavengers, but not the scavengers of superoxide or a MnSOD mimetic, reduced the release of cytochrome c and abolished the sensitization of the cells to cisplatin-induced apoptosis. Such inhibition of ROS prevented the processing and release of AIF (apoptosis-inducing factor) and HTRA2 from mitochondria. Furthermore, suppression of autophagy in NSCLC cells with active basal autophagy reduced their proliferation without significant effect on the cell-cycle distribution. Inhibition of cell proliferation delayed accumulation of cells in the S phase upon treatment with etoposide that could attenuate the execution stage of etoposide-induced apoptosis. These findings suggest that autophagy suppression leads to inhibition of NSCLC cell proliferation and sensitizes them to cisplatin-induced caspase-dependent and -independent apoptosis by stimulation of ROS formation.
Keywords: NSCLC; ROS; apoptosis; autophagy; caspase-independent cell death; hydroxyl radical; superoxide.
Figures
References
-
- Pyo JO, Nah J, Kim HJ, Lee HJ, Heo J, Lee H, et al. Compensatory activation of ERK1/2 in Atg5-deficient mouse embryo fibroblasts suppresses oxidative stress-induced cell death. Autophagy. 2008;4:315–21. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical